1. Arbres et graphes en R
Comment définit-on des arbres et des graphes en R ?
Application : générer les structures et reproduire les graphiques suivants.
Pour le graphe, la liste des arcs est dans le fichier list_arcs.txt ; pour l'arbre, on peut utiliser le format de dot, soit le fichier arbre.dot.txt.
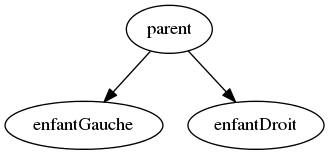
Pour l'arbre, on peut se dispenser de lire le fichier, vu qu'il n'y a que deux arcs et que le package igraph dispose d'une fonction graph.formula() :
# tracé d'un arbre minimal avec le package igraph
require(igraph)
g <- graph.formula(parent-+enfantGauche,parent-+enfantDroit)
# tracé approximatif
plot.igraph(g,vertex.size=70,layout=layout.lgl)
# tracé plus équilibré via les coordonnées x et y
# et sans couleur
V(g)$color <- NA
plot.igraph(
g,
vertex.size=70,
layout=cbind(c(200,100,300),c(300,100,100))
) # fin de plot.igraph
# lecture du fichier arbre.dot.txt
# dont le contenu est
#
# parent -> enfantGauche ;
# parent -> enfantDroit ;
arb <- read.table("arbre.dot.txt")
# un peu de ménage et on transforme en graphe
arb$V2 <- NULL
arb$V4 <- NULL
g <- graph.data.frame(arb)
# et voilà !
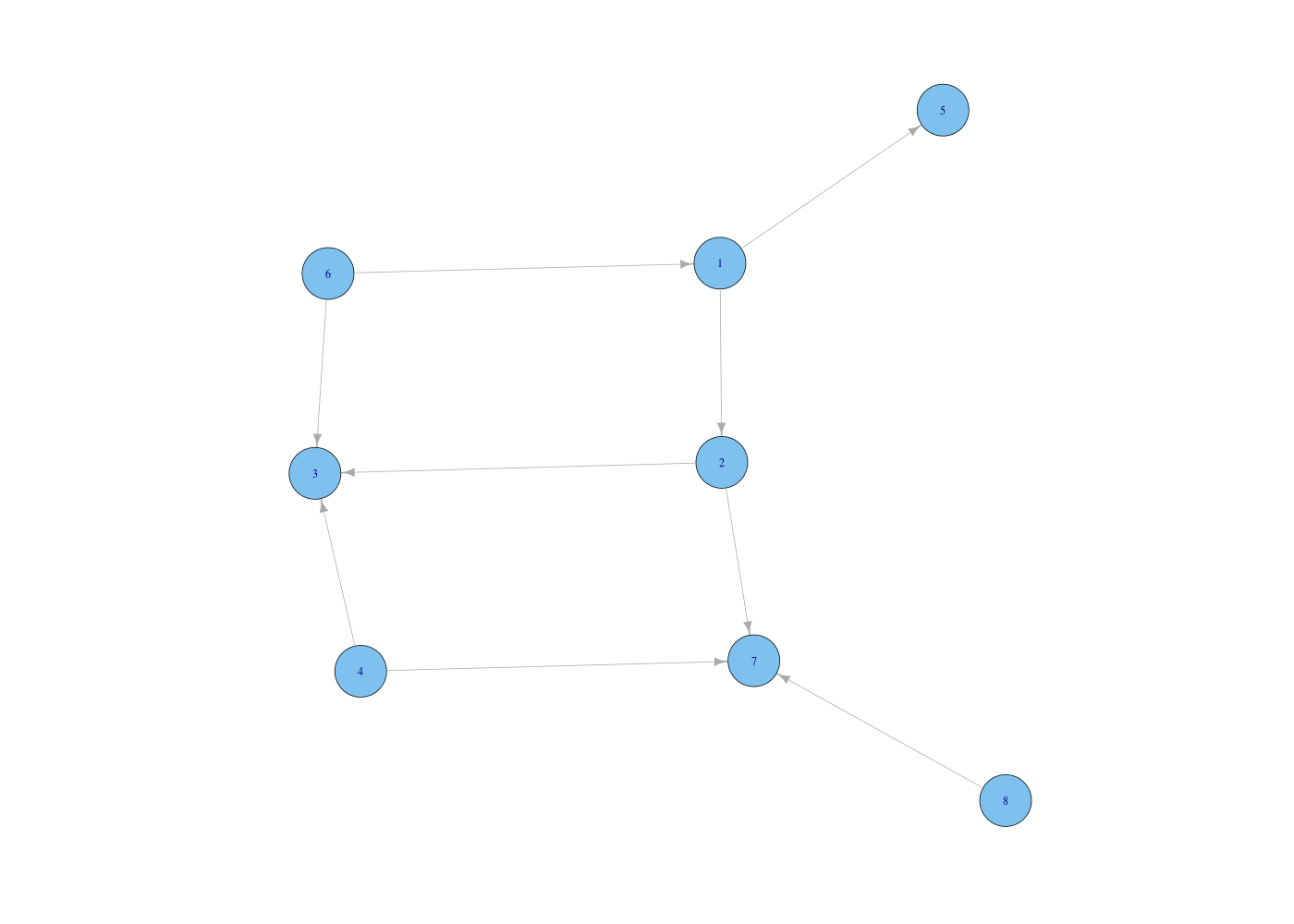
Pour le graphe, le fichiers des arcs est dans un format reconnu par la fonction read.graph() donc le lire et le tracer est très simple :
# tracé d'un graphe avec le package igraph
# les arcs du graphe sont définis ligne par ligne
# début du fichier :
# 0 1
# 0 4
# 1 2
# ...
# lecture du graphe
gra <- read.graph("list_arcs.txt",format="edgelist")
# tracé du graphe
plot.igraph(gra)
D'autres packages R intéressants à consulter pour les graphes : ggraph, introduction à ggraph et tidygraph, introduction à tidygraph sans oublier la Task View nommée gR.
2. Tableaux associatifs et dictionnaires
Comment construire les dictionnaires alphabétiques et fréquentiels d'un texte en R ? On utilisera des tableaux associatifs où les clés sont les mots et les valeurs leur nombre d'occurrences.
Application : analyser les deux premiers chapitres du Candide de Voltaire disponibles dans le fichier candide.txt.
Nos fonctions lit.texte et analexies sont prévues pour cela, qui utilisent le package hash :
# chargement des fonctions
source("http://forge.info.univ-angers.fr/~gh/wstat/statgh.r",encoding="latin1")
# lecture et construction des dictionnaires
dicos <- analexies( lit.texte("candide.txt",encoding="latin1") )
# affichage court
print (str(dicos))
# pour vérification, début des dictionnaires
cat("Mots\n")
print( head(dicos$tmots) )
cat("Occurences\n")
print( head(dicos$toccs) )
1958 mots en tout dont 731 mots distincts.
chaque mot est donc répété 2.68 fois en moyenne.
List of 5
$ nmots : num 1958
$ hmots :Formal class 'hash' [package "hash"] with 1 slot
.. ..@ .xData:<environment: 0x519f3d8>
$ nmotds: num 731
$ tmots : num [1:731, 1] 1 1 1 1 1 1 1 1 1 1 ...
..- attr(*, "dimnames")=List of 2
.. ..$ : chr [1:731, 1] "1" "10" "11" "12" ...
.. ..$ : chr "occ"
$ toccs : num [1:731, 1] 77 72 61 45 44 42 38 29 28 25 ...
..- attr(*, "dimnames")=List of 2
.. ..$ : chr [1:731] "de" "et" "le" "la" ...
.. ..$ : chr "occ"
NULL
Mots
occ
1 1
10 1
11 1
12 1
13 1
14 1
Occurences
occ
de 77
et 72
le 61
la 45
candide 44
il 42
3. Alignements, phylogénie, analyse d'images
Comment lit-on des séquences Fasta en R ? Comment les aligne-t-on (qu'est-ce ?) ? Comment en construit-on une phylogénie (qu'est-ce ?) ?
Application : aligner les protéines de la classe 3 de la LEADPB et en donner un arbre phylogénétique. Où est le problème ? On pourra utiliser le fichier classe3.fasta.
Lire des séquences Fasta peut se faire avec la fonction read.fasta() du package seqinr :
# lecture de classe3.fasta via read.fasta du package seqinr
require(seqinr)
cl3 <- read.fasta("classe3.fasta",seqtype="A")
print( head(cl3) )
$AAA99962_cl_3
[1] "M" "A" "G" "V" "I" "H" "K" "I" "G"...
[80] "Q" "Q" "Y" "R" "G" "G" "E" "H" "R"...
[159] "K" "K" "K" "E" "K" "K" "K" "H" "E"
attr(,"name")
[1] "AAA99962_cl_3"
attr(,"Annot")
[1] ">AAA99962_cl_3"
attr(,"class")
[1] "SeqFastaAA"
$AAA99963_cl_3
[1] "M" "A" "G" "V" "I" "H" "K" "I" "G"...
[80] "G" "G" "E" "K" "K" "K" "K" "E" "K"
attr(,"name")
[1] "AAA99963_cl_3"
attr(,"Annot")
[1] ">AAA99963_cl_3"
attr(,"class")
[1] "SeqFastaAA"
$AAC05356_cl_3
[1] "M" "A" "G" "I" "I" "N" "K" "I" "G"...
[80] "E" "G" "G" "A" "G" "E" "K" "K" "K"
attr(,"name")
[1] "AAC05356_cl_3"
attr(,"Annot")
[1] ">AAC05356_cl_3"
attr(,"class")
[1] "SeqFastaAA"
$AAC25776_cl_3
[1] "M" "A" "G" "I" "M" "N" "K" "I" "G"...
[80] "D" "H" "K" "E" "G" "Y" "H" "G" "E"
attr(,"name")
[1] "AAC25776_cl_3"
attr(,"Annot")
[1] ">AAC25776_cl_3"
attr(,"class")
[1] "SeqFastaAA"
$AAK52077_cl_3
[1] "M" "S" "G" "V" "I" "H" "K" "I" "G"...
[80] "G" "E" "H" "R" "E" "G" "E" "H" "K"
attr(,"name")
[1] "AAK52077_cl_3"
attr(,"Annot")
[1] ">AAK52077_cl_3"
attr(,"class")
[1] "SeqFastaAA"
$AAN37899_cl_3
[1] "M" "A" "G" "I" "I" "H" "K" "I" "E" "...
[81] "S" "S" "S" "D" "S" "D"
attr(,"name")
[1] "AAN37899_cl_3"
attr(,"Annot")
[1] ">AAN37899_cl_3"
attr(,"class")
[1] "SeqFastaAA"
Malheureusement ceci ne nous aide pas beaucoup parce qu'il faut disposer d'un alignemnent comme celui-là avant d'utiliser la fonction read.alignment() du package seqinr :
# lecture de l'alignment de la classe3 via read.alignment du package seqinr
require(seqinr)
cl3_ali <- read.alignment("classe3_ali.fasta",format="fasta")
print( head(cl3_ali) )
# matrice des distances
cl3_dist <- dist.alignment(cl3_ali, matrix = "identity")
# classification
cl3_hie <- hclust(cl3_dist)
# tracé
plot(cl3_hie)
[Affichage tronqué]
$nb
[1] 35
$nam
[1] "AAA99962_cl_3 179 Weight: 0.69" ...
[8] "AGG55635_cl_3 131 Weight: 0.88" ...
[15] "AAA99963_cl_3 106 Weight: 1.15" ...
[22] "AER13138_cl_3 112 Weight: 0.97" ...
[29] "AAT81473_cl_3 107 Weight: 1.16"
$seq
$seq[[1]]
[1] "magvihkig---ealhvgggqkeedkskg----ehqs--
$seq[[2]]
[1] "msgvihktg---ealhmgggqkeedkhkg----ehhs--
$seq[[3]]
[1] "msgvihkig---ealhmgggqkeedkhka----ehhs--
$seq[[4]]
[1] "msgvihkig---ealhmgggqkeedkhkg----ehhs--
$seq[[5]]
[1] "msgvihktg---ealhmgggqkeedkhkg----ehhs--
$seq[[6]]
[1] "magimnkig---dalhgggdkkege-hkg----eqhghv-
$seq[[7]]
[1] "magimnkig---galhiggdkkege-hkg----eqhghv-
[1] "maniihkie---etlgmggdkh---ke------gdq-----
$com
[1] NA
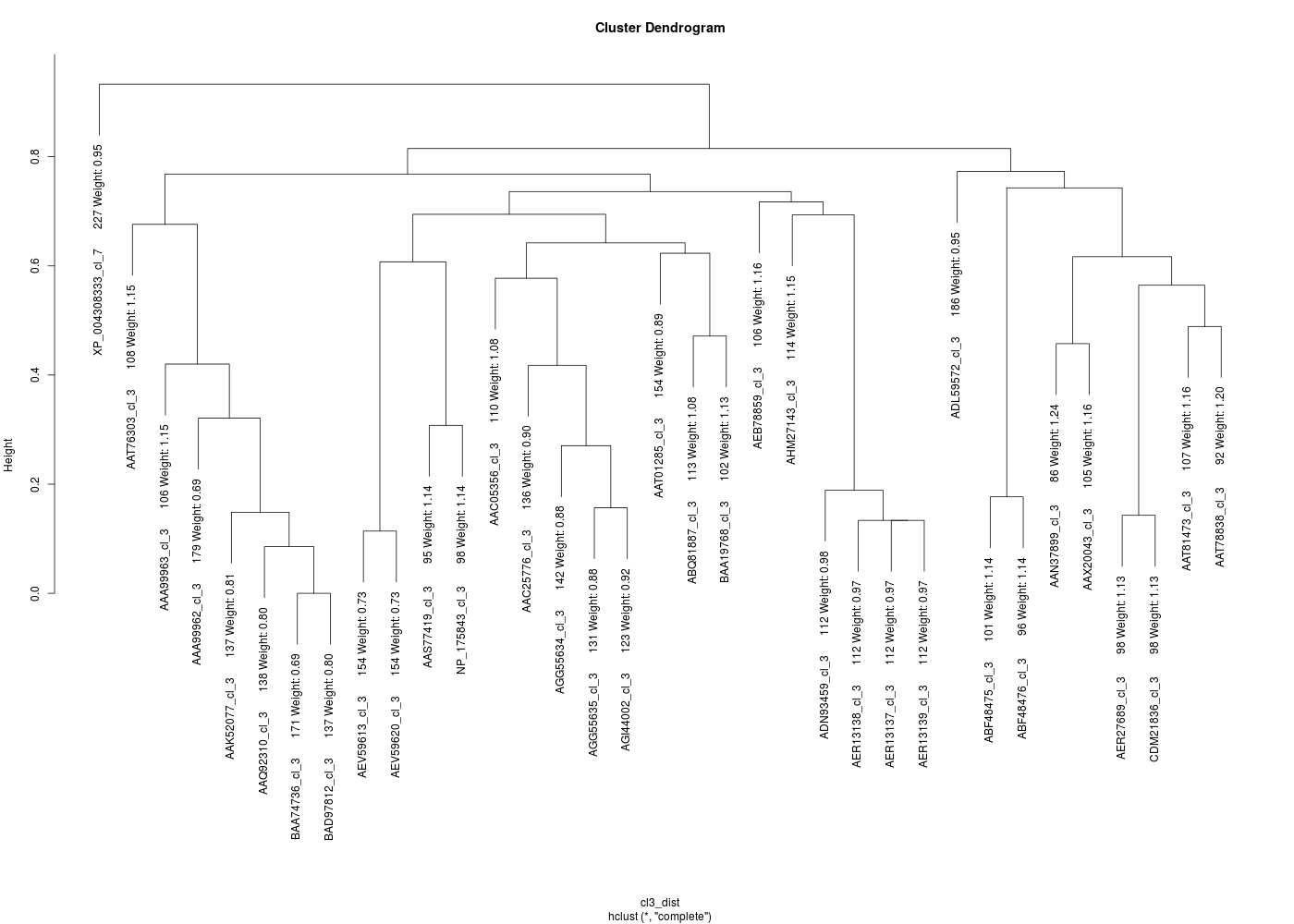
Il n'est malheureusement pas possible d'expliquer en trois phrases ce qu'est une phylogénie, ni une classification hiérarchique. Que R soit capable de calculer et tracer ces objets en trois instructions est un autre problème...
4. Gènes différentiellement exprimés et clustering
Quel est le lien entre R et BioConductoR ?
Comment fait-on pour trouver les gènes différentiellement exprimés issus d'un séquençage ?
Attention : il ne s'agit pas ici de programmation mais d'utilisation d'outils programmés en R. Cela demande d'abord de la culture en biologie (gène) et en statistiques (différentiellement exprimés) plus une bonne dose de bioinformatique. Pour s'en convaincre, la réponse à ce genre de quesions est dans ce genre d'ouvrages :
Pour une introduction rapide à Bioconductor et des exemples d'analyses avec Bioconductor, on pourra notamment lire en anglais Bioconductor-tutorial.pdf et, en français, TP_Affy.pdf disponibles ici et là en version locale.
5. Elimination de variables transitivement corrélées
Il arrive qu'on ait un tableau de données avec beaucoup de colonnes quantitatives. En statistiques, on apprend à ne pas conserver des variables trop ressemblantes ("corrélées"). Ecrire une fonction qui à partir d'un tableau de données trouve les groupes de variables qui se ressemblent transitivement (si A ressemble à B et B ressemble à C, alors A ressemble à C) et qui ne conserve qu'une variable par groupe.
Application : essayer de définir des groupes de variables pour les données 710x46.dar.
Code-source php de cette page. Retour à la page principale du cours.
|


 Retour à la page principale de
(gH)
Retour à la page principale de
(gH)