![]()
Présentation du projet
Résumé :
Le but du projet est de calculer des distances entre les divers "objets" classiques de la bioinformatique :
- chaines de caractères, - séquences nucléotidiques (ADN), - séquences polypeptidiques (AA), - arbres [binaires enracinés], - matrices [de distances]. Ces distances seront implémentées sous la forme de sous-programmes PHP 5 classiques puis comme des méthodes objets si le temps le permet. Elles seront utilisées à titre de démonstration dans une page Web avec des formulaires de saisie des objets et devront pouvoir servir à calculer des matrices de distances en ligne de commande.
Détail du projet:
Quelques références Web utiles :Une distance est une fonction mathématique qui associe à deux élements une valeur numérique positive ou nulle qui quantifie la proximité (ou l'éloignement) entre ces éléments. En bioinformatique, on parle plutot volontiers d'objets que d'éléments c'est pourquoi nous utiliserons l'expression "distance entre objets".
La bioinformatique utilise de nombreuses distances entre "séquences biologiques" qu'il s'agisse de séquences adn ou de séquences d'acides aminés. Ces séquences, alignées ou non, sont des chaines de caractères particulières pour lesquelles il existe déjà en informatique des distances classiques (distance de Hamming, distance d'édition...). Il faudra donc commencer par recenser et programmer ces distances classiques avant de programmer les distances spécifiques aux séquences bioinformatiques.
Deux autres grandes familles d'objets pour lesquelles il est utile de savoir calculer des distances en bioinformatique sont les matrices et les arbres. S'il existe de nombreuses distances relativement faciles à comprendre et à programmer pour les matrices, il n'en va pas de même pour les arbres (mais on peut associer une "matrice de distances" à un arbre pour contourner le problème).
Programmer les distances comme des fonctions est assez simple. Par contre, en faire des méthodes-objet et définir les objets associés est plus délicat. C'est pourquoi cette partie du projet ne sera effectuée que si tout le reste est réalisé et documenté.
Dans le cadre du projet, les fonctions programmées seront utilisées dans deux contextes différents. Le premier contexte sera celui d'une page Web où on pourra soit utiliser des objets préparés, soit les saisir à l'aide de formulaires. La page Web permettra de choisir quelle(s) distance(s) on veut utiliser. Le deuxième contexte sera celui d'un programme en ligne de commande où le but est de calculer toutes les distances entre les objets définis par les lignes d'un fichier-texte dont on donne le nom en paramètre.
On réalisera le projet en Php qui devra s'éxécuter aussi bien sous Unix que sous Windows, (que l'on testera pour php 5) avec l'option d'exécution error_reporting(E_ALL).
distance définition wiki levenshtein la distance de levenshtein existe déjà en php. distance d'édition programmation dynamique (voir surtout demo1.pdf). arbres phylogénétiques voir surtout les transparents de l'exposé. oop5 objets en php5 (manuel php) exemple d'interface avec exemples préparés
Fichiers de données pour tester les distances
desmots.txt C'est un fichier de mots de la langue française pour lesquels les distances classiques sont faciles à calculer.j6_seq.txt
Contenu :M01 a MO2 ah M03 bah M04 bac M05 banc M06 bien M07 avion M08 avions M09 bravo M10 zèbre M11 êtreC'est un fichier des séquences Fasta (acides aminés) des protéines ; chaque séquence est représentée par deux lignes : la première contient le symbole > puis l'identifiant PDB de la protéine puis le symbole : et enfin la lettre associée à la chaine ; la seconde, éventuellement très longue, contient la liste des acides aminés. Les séquences sont séparées par une ligne vide.j6_codons.txt
Extrait du fichier :>1DKL:A QSEPELKLESVVIVSRHGVRAPTKATQLMQDVTPDAWPTWPVKLGWLTPRGGELIAYLGHYQRQRLVADGLL... >1APH:A GIVEQCCASVCSLYQLENYCN >1APH:B FVNQHLCGSHLVEALYLVCGERGFFYTPKA >1VKT:A GIVEQSCTSISSLYQLENYCN >1VKT:B FVNQHLCGSDLVEALYLVCGERGFFYTKPTC'est un fichier des codons (acides nucléotidiques). On trouve éventuellement des commentaires repérés par le symbole # comme premier caractère non nul de ligne. Les protéines sont séparées par une ou plusieurs lignes vides. Chaque protéine comporte une ou plusieurs séquences de codons. Une séquence de codons est écrite sur une seule ligne, éventuellement très longue selon le format suivant : l'identifiant PDB de la protéine, le symbole :, la lettre associée à la chaine, le symbole |, une indication de correspondance swissport (à ignorer), un ou plusieurs espaces, les nucléotides.arbre1
Extrait du fichier :# j6_codons.txt ; le 21/07/04 vers 03:56:55 extraction du 01/06/05 vers 17:05:12 1PS3:A|CAA54732p63 aacattgagaacaagctgcatgagctggaaaa... 1APH:A|AAA30722p84 gaggtggagggcccgcaggtgggggcgctggagctggccggaggcccgggcgcgggcggcctg 1APH:B|AAA30722p24 tggcccccccccccggcccgcgccttcgtcaa...Il s'agit d'un arbre binaire indicé (d'où le suffixe .abi) construit sur 6 séquences (non fournies) nommées A, B... F. On trouve les éléments au format numéro/nom puis les noeuds au format numéro/indice de niveau/numéro ainé/numéro benjamin.arbre2
Contenu :
1 A 2 B 3 C 4 D 5 E 6 F 7 2 1 2 8 4 3 7 9 6 4 5 10 6 8 9 11 8 6 10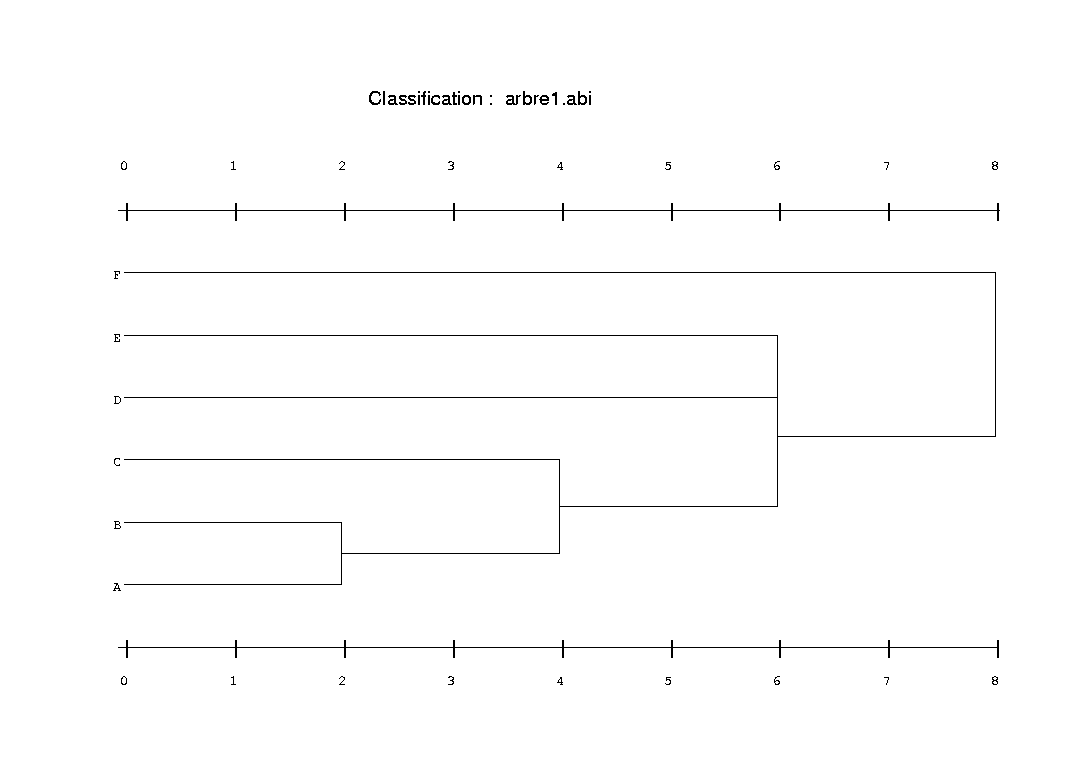 Il s'agit d'un autre arbre binaire indicé (d'où le suffixe .abi) construit sur les mêmes 6 séquences que arbre1.matrice1
Il s'agit d'un autre arbre binaire indicé (d'où le suffixe .abi) construit sur les mêmes 6 séquences que arbre1.matrice1
Contenu :
1 A 2 B 3 C 4 D 5 E 6 F 7 1 3 1 8 4 2 5 9 6 4 7 10 6 8 9 11 8 6 10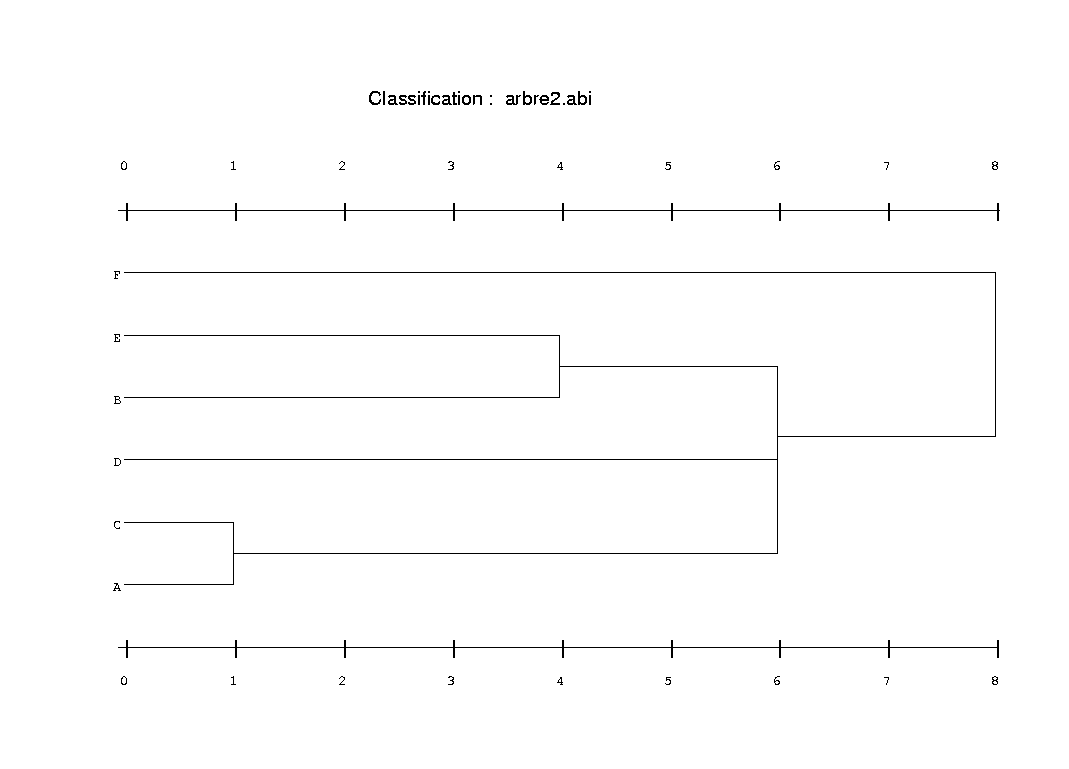 Cette matrice est "la" matrice "canonique naturelle" associée à arbre1.matrice2
Cette matrice est "la" matrice "canonique naturelle" associée à arbre1.matrice2
Contenu :A B C D E F A 0 B 2.0000 0 C 4.0000 4.0000 0 D 6.0000 6.0000 6.0000 0 E 6.0000 6.0000 6.0000 6.0000 0 F 8.0000 8.0000 8.0000 8.0000 8.0000 0Cette matrice est "la" matrice "canonique naturelle" associée à arbre2.
Contenu :A B C D E F A 0 B 6.0000 0 C 1.0000 6.0000 0 D 6.0000 6.0000 6.0000 0 E 6.0000 4.0000 6.0000 6.0000 0 F 8.0000 8.0000 8.0000 8.0000 8.0000 0
Retour à la page principale de (gH)