![]()
![]()
Conseil Scientifique METASEED : axe 4(mardi 8 décembre 2015)
Table des matières cliquable
1. Rappel de l'axe 4
2. Collecte des données
3. Automatisation de l'exploitation
4. Bilan
|
 Retour à la page principale de
(gH)
Retour à la page principale de
(gH)


Conseil Scientifique METASEED : axe 4
(mardi 8 décembre 2015)
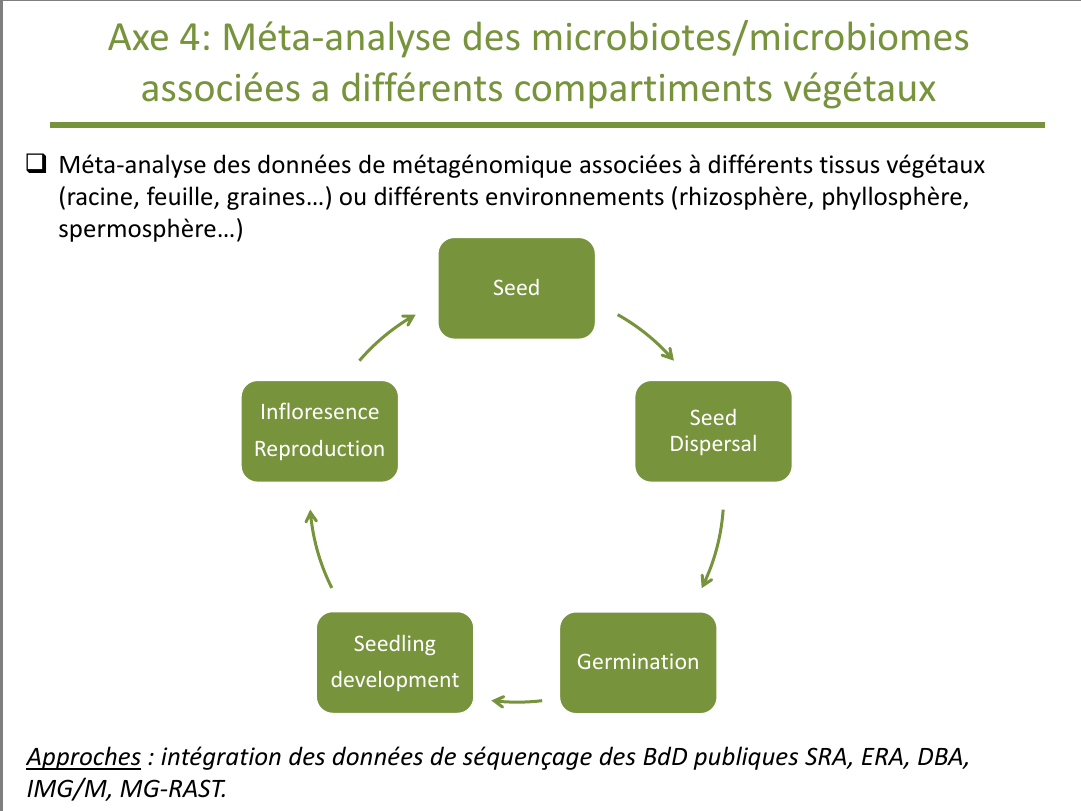
Table des matières cliquable
3. Automatisation de l'exploitation
4. Bilan
1. Rappel de l'axe 4
Dans l'axe 4 il est prévu de réaliser des méta-analyses au niveau des différents compartiments dans chacun des stades du développement végétal. La première étape, essentielle, est donc la collecte des données, alors que la deuxième étape, suite logique, en est le traitement.
2. Collecte des données
Il existe dans le monde trois grandes bases de données susceptibles de nous intéresser : SRA (the NCBI's Sequence Read Archive -- NGS data), ENA (the EMBL's European Nucleotide Archive), DBA (the DDBJ's Sequence Read Archive).
Malheureusement pour nous, ces bases sont plus prévues pour le dépot de séquences que pour une recherche «intelligente« de données. De plus ces bases sont sans doute miroirs les unes des autres, mais avec leur propre système de numérotation des study, run, experiment.
Comme souvent dans une base de données, le plus simple -- pour un(e) informaticien(ne) -- est de connaitre l'identifiant de ce qu'on cherche, ce qui bien sûr est une tache difficile voire ingrate pour un utiisateur de base. Cliquer sur l'image ci-dessous pour voir comment on navigue dans SRA :
Une première tentative, initiée dans le cadre du stage de M2 de QuRP, a été de rechercher des mots-clés dans une vingtaine de publis puis d'interroger les bases de données avec ces mots-clés. Cette approche est moyennement satisfaisante dans la mesure où il y avait beaucoup de faux positifs renvoyés avec ces termes.
Nous avons donc tenté une autre approche qui consiste à trouver directement ces numéros d'accession, comme PRJNA208116, PRJEB5860 ou SRR1524567 dans des articles sélectionnés par MaBA. Voici deux des articles choisis :
Structure and function of the bacterial root microbiota in...
Metaproteogenomic analysis of microbial communities in the phyllosphere and...
Pour éviter de passer trop de temps à lire nous-mêmes les articles (ce qui s'automatise mal), nous avons essayé de détecter l'environnement de mots comme database, accession, number. Et effectivement, il est assez simple d'extraire des articles des informations comme
[Bulgarelli_2015.txt] European Nucleotide Archive (ENA) under the accession number PRJEB5860. [Knief_2012.txt] under accession numbers HE589809 to HE589931. [Knief_2012.txt] PRIDE database under accession number 1689. [Maignien_2014.txt] Nucleotide sequence accession number. All sequences described have been [Ofek-Lalzar_2014.txt] in the NCBI Sequence Read Archive under BioProject accession number PRJNA208116,Une fois ces numéros d'accession trouvés (ou pas!), il faut savoir à quelles données ils correspondent. Heureusement, le logiciel R fournit un package nommé SRAdb dont la prise en main est assez facile... une fois les descripteurs de la base chargée, ce qui prend un certain temps car il faut rapatrier 15 Go de métadonnées. Les champs (ou colonnes) de la base de données SRA ont des noms explicites comme on peut le voir ci-dessous :
Il ne reste plus qu'à écrire un petit programme en R pour interroger ces tables de données avec ces numéros, soit les paramètres :
Mais il ne faut pas crier victoire trop vite : les metadata sont souvent mal renseignés voire non renseignés. Voici des exemples de ce qu'on trouve comme renseignements censés nous aider à comprendre ce que contiennent les données
De plus il faut trouver la taille des données et non pas tout télécharger. Heureusement, nous avons finalement réussi à trouver une option du logiciel wget qui donne en ligne de commandes la taille d'un fichier dont on passe l'URI, comme par exemple ici la taille 12688427 -- soit environ 13 Mo -- pour le fichier SRR1524567.sra.
wget --spider ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/ reads/ByExp/sra/SRX/SRX660/SRX660006/SRR1524567/SRR1524567.sra Résolution de ftp-trace.ncbi.nlm.nih.gov (ftp-trace.ncbi.nlm.nih.gov)... 130.14.250.13, 2607:f220:41e:250::13 Connexion vers ftp-trace.ncbi.nlm.nih.gov (ftp-trace.ncbi.nlm.nih.gov) 130.14.250.13|:21... connecté. Ouverture de session en anonymous... Session établie! ==> SYST ... complété. ==> PWD ... complété. ==> TYPE I ... complété. ==> CWD (1) /sra/sra-instant/reads/ByExp/sra/ SRX/SRX660/SRX660006/SRR1524567 ... complété. ==> SIZE SRR1524567.sra ... 12688427 ==> PASV ... complété. => «.listing» ==> CWD (1) /sra/sra-instant/reads/ByExp/sra/ SRX/SRX660/SRX660006/SRR1524567 ... complété. ==> PASV ... complété. ==> LIST ... complété. Le fichier «SRR1524567.sra» existe.Pour les autres bases, pas de package, pas de fonction et donc soit on attend un hypothétique package R soit il faudra écrire un programme R pour interfacer les base des données... Ainsi, pour le numéro ERP001384, il est possible de consulter les informations en ligne dans un navigateur, à l'aide du lien ci-dessous
https://www.ebi.ac.uk/ena/data/search?query=ERP001384
Ceci s'obtient facilement si on tape ce numéro d'accession dans le panneau Search de l'ENA

Mais entre parcourir des informations dans une page Web et construire un fichier de résultats, il y a un un grand travail à fournir...
Au final, de façon sûre et automatique, voici ce que nous avons obtenu pour 26 articles sélectionnés
Et voici ce que nous avons "bricolé" en semi-automatique avec une intervention humaine :
[ Le fichier complet des mots est ici. ]
Il est clair que ce n'est pas très satisfaisant pour décider si ces données sont pertinentes pour nos études dans l'axe 4.
3. Automatisation de l'exploitation
Nous avions présenté l'an dernier un script Perl nommé metauto16S disponible sur le Web avec une page web d'explication et dont le but était d'analyser des données de séquençage amplicon de façon automatisée. Moyennant un "petit enrobage" réalisé par MaBR, ingénieur à la station, ce script est désormais disponible dans la plate-forme Galaxy.
Ce script a également été adapté pour le gène gyrb.
Suite au stage de M2 de QuRP, nous disposons d'un nouvel ensemble de commandes pour traiter des données de shotgun et dont l'ensemble est presque utilisable par un utilisateur "moyen".
4. Bilan
Le bilan est, pour l'instant, «mitigé» : si la partie mise en place de scripts d'exécution une fois les données rapatriées est bien avancée (un utilisateur "moyen" peut presque s'en servir tout seul, en ligne de commandes ou via Galaxy), la récupération des données est plus problématique, et ce, à deux titres :
retrouver des données via des numéros d'accession est beaucoup moins simple que prévu ;
la qualité des méta-données -- qui doit servir à sélectionner les données -- est très décevante et les données sont parfois inexploitables faute de renseignements associés.