Conseil Scientifique METASEED : axe 4
(mercredi 10 décembre 2014)
Table des matières cliquable
1. Présentation générale de l'axe 4
2. Bases de données utilisées
3. Quelles analyses ? quels termes choisir ? quels outils utiliser ?
4. Présentation du script perl metauto
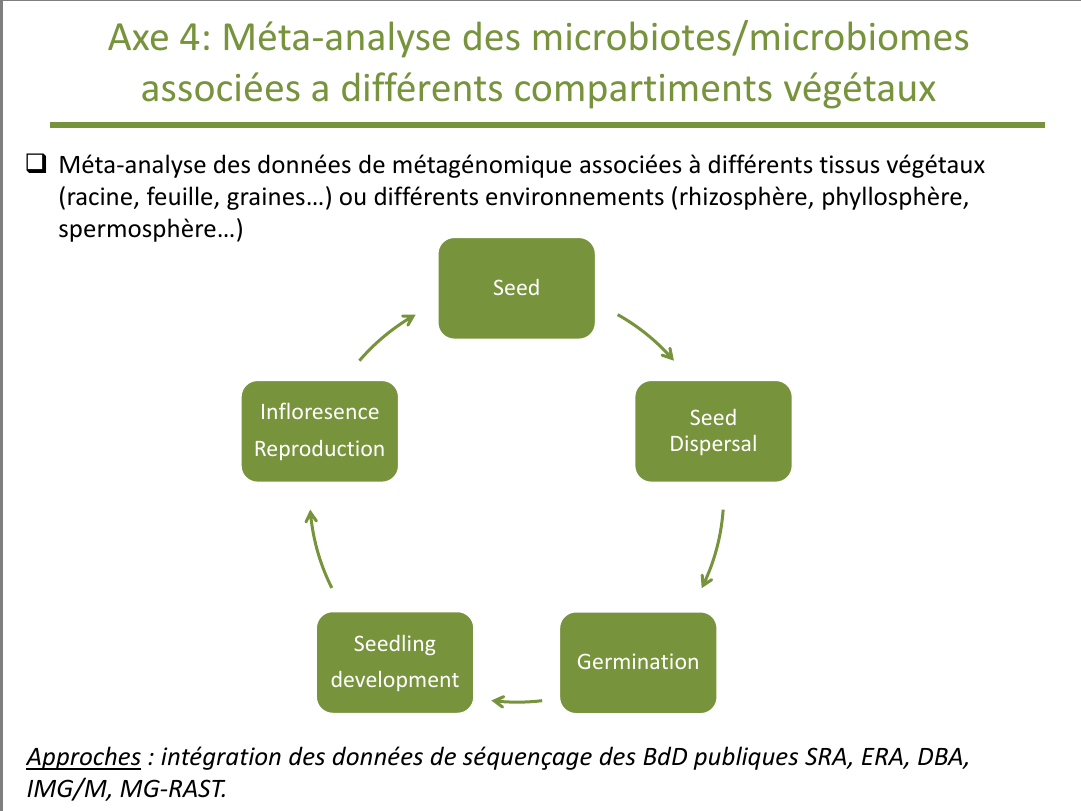
S. REZKI et M. BARRET ont présenté les résultats des travaux sur semences
et plantules.
Au cours du dernier volet du projet metaSEED, nous souhaitons intégrer
des données de séquençage effectué sur l'ensemble des tissus végétaux (graine, racine...)
et leurs environnements (spermosphère, rhizosphère...)
aux séquences obtenues lors des axes opérationnels précédents
sur l'ensemble du développement végétal (dispersion, germination, inflorescence...) afin de réaliser des méta-analyses
au niveau des différents compartiments dans chacun des stades de ce développement.
| Tissu |
Environnement |
| graine |
spermosphère |
| racine |
rhizosphère |
| feuille |
phyllosphère |
| fleur |
anthosphère |
| fruit |
carposphère |
Par exemple, certains laboratoires ont récemment publié des travaux
de recherche sur le microbiote des racines d' A. thaliana
et d'autres groupes ont engagé des études métagénomiques de la phyllosphère des Brassicaceae, d'autres sur la rhizosphere...
Quelques exemples de publications récentes correspondantes avec lien PubMed :
La méta-analyse des microbiotes et des métagénomes devrait permettre
d'apporter des éléments de réponse sur le rôle de la graine en tant que source
d'inoculum pour la plante et les liens fonctionnels existant entre ces
différents écosystèmes, même si nous sommes bien conscients
que le sol et son écosystème joue une part importante dans ces interactions, part qu'il est difficile d'évaluer et d'analyser.

Qui dit méta-analyse dit analyse de séries d'essais et de résultats. Comme nous
n'allons pas réaliser toutes ces essais, nous allons aller chercher les données sur le web.
Donc pour réaliser les méta-analyses, nous allons nous appuyer sur
au moins trois grandes bases de données internationales qui sont heureusement
mirroir les unes des autres : SRA, ENA et DRA.
| SRA |
The NCBI's Sequence Read Archive (NGS data). |
| ENA |
EMBL's European Nucleotide Archive. |
| DBA |
DDBJ's Sequence Read Archive. |
Mais il existe aussi deux autres bases de données plus spécialisées,
comme MG-RAST et IMG.
| mg-rast |
The MetaGenomic RAST |
| img/m |
Integrated Microbial Genomes with Microbiomes samples |
Dans SRA/ENA/DRA on trouve
principalement des reads c'est-à-dire des données brutes alors que dans MG-RAST/IMG on trouve
des données déjà un peu analysées sous forme de contigs.
Même si on trouve les mêmes données dans SRA/ENA/DRA, nous allons nous focaliser sur SRA parce qu'il
assez simple d'interfacer SRA (c'est-à-dire d'y accéder par programme). Par exemple
en utilisant le logiciel R et son package nommé SRAdb
dont le manuel de
présentation est
ici et le manuel de référence
là.

|

|
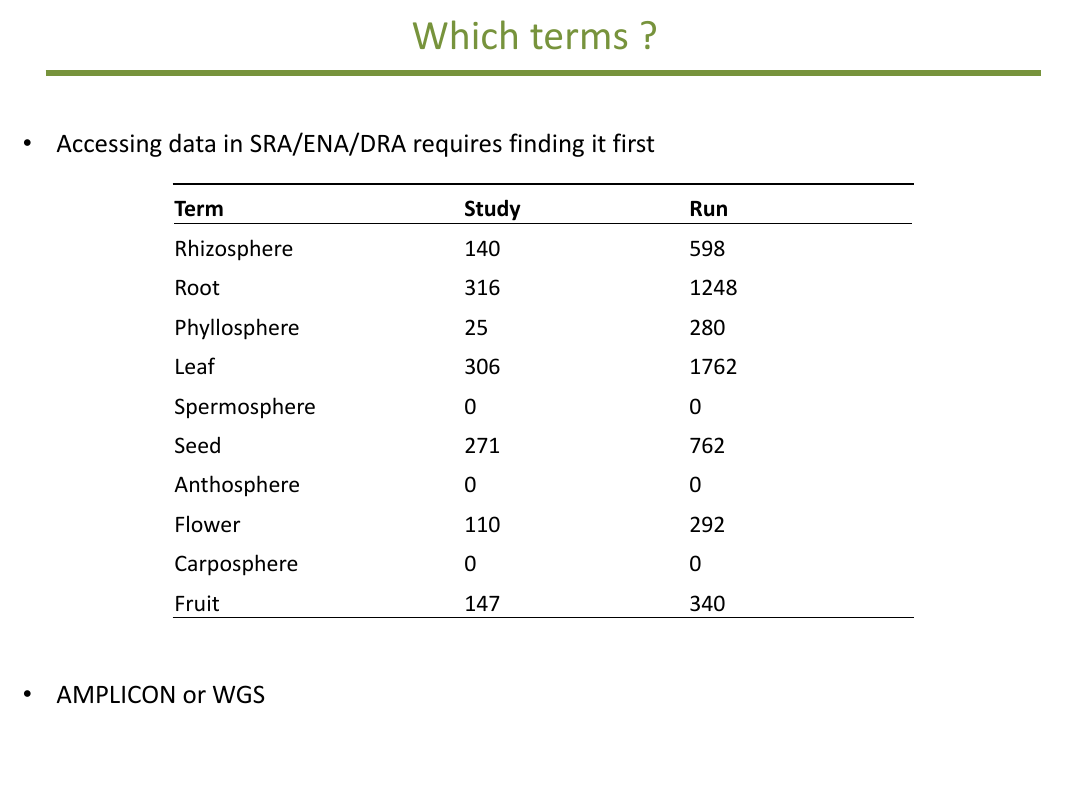
Comment procéder ? Nous voulons rapatrier des données et les analyser selon
un même protocole. Pour récupérer des données sans crouler sous les «big giga of data»,
il va falloir chercher des termes précis dans ces bases de données.
C'est là que les difficultés commencent. Lorsqu'on dépose des données, on doit renseigner de nombreux champs pour information. Le terme study, par exemple, signifie étude dynamique, analyse générale alors que run représente seulement les résultats d'un seul séquençage -- ce qui peut inclure plusieurs échantillons. Ainsi M. BARRET a déjà déposé un run au sein d'ENA soit 252 échantillons (samples) dont chacun contient 2 fichiers fastq repérés par R1 et R2. Comme chaque fichier fastq contient entre 10 000 et 50 000 reads cela fait déjà beaucoup de données. En médiane : 5 millions de reads (liste/description des fichiers). Nombre de fichiers pour 16S : 172, de taille moyenne 47 Mo soit une taille totale de 8,2 Go (1,5 Go une fois compressés).
Si nous savons ce que nous cherchons (racine et rhizosphère, fruit et carposphère...), nous ne savons
pas comment les autres ont écrit ces termes. Ainsi faut-il chercher leaf et leaves ? Comment filtrer
("à la main" ? par programme ?) les résultats quand il y en a plusieurs milliers ?
Il faut aussi faire des choix de type d'analyses. Lorsque les données déposées sont des simples reads
on peut réaliser des analyses d'amplicons ou de génomes entiers, ce qui nous mène à des analyses WGS (Whole Genome Shotgun) et non pas à des analyses GWAS. Les valeurs indiquées dans le tableau à droite correspondent aux nombres de résultats trouvés par une recherche
manuelle pour fin novembre 2014.
|

|
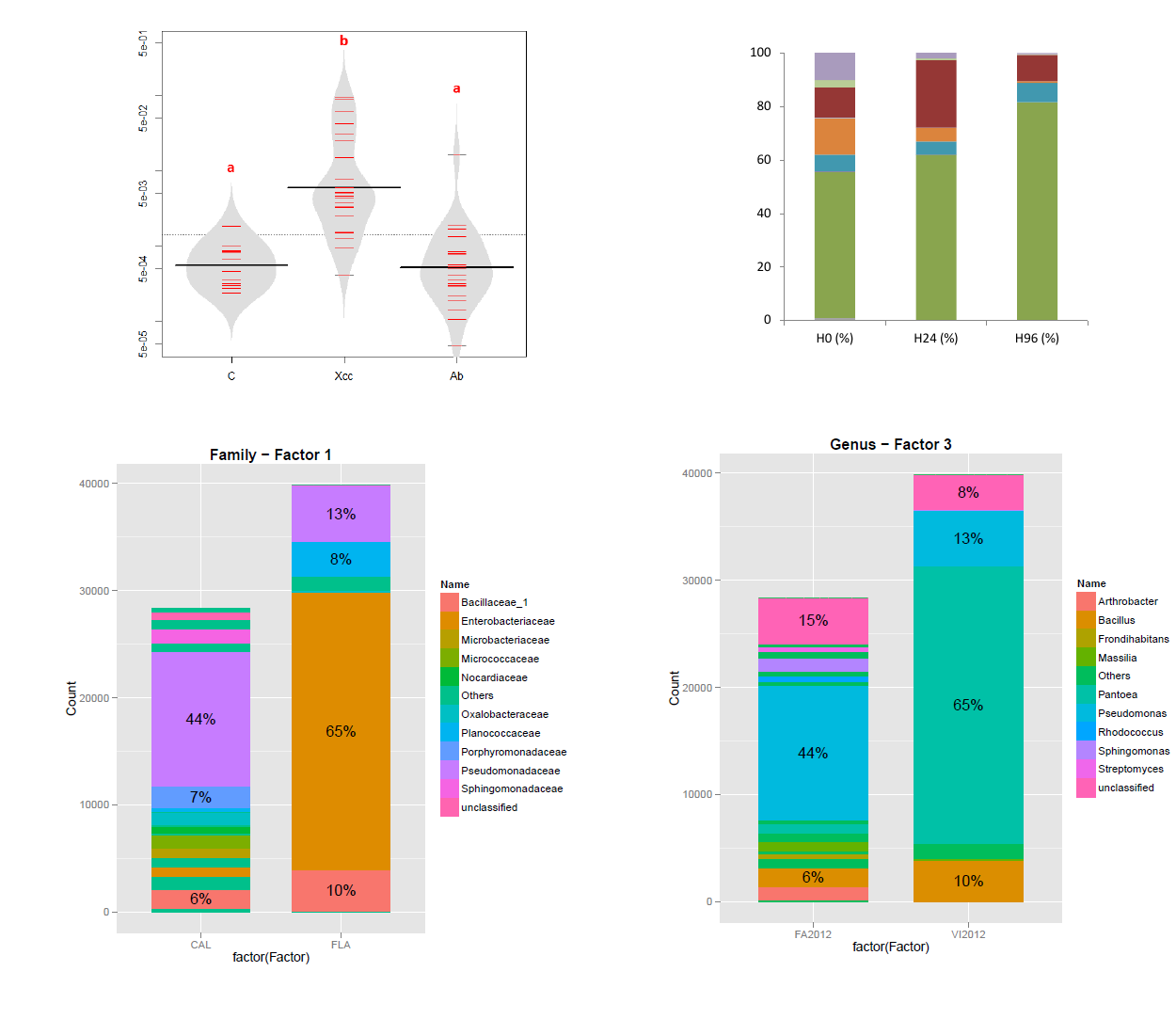
Pour les analyses de type amplicon 16S, nous avons déjà mis au point courant 2014 un script écrit en perl nommé
metauto (METAgenomic AUTOmatic analysis) qui ne demande en entrée que des reads et une arborescence
précise avec trois répertoires nommés Input/, Output/ et Results/.
Ce script est disponible sur internet et expliqué via une page Web dédiée. Nous allons l'étendre aux autres analyses de type amplicon.
Si la durée d'exécution du script en fonction des données est long (de quelques minutes à plusieurs
heures, voire quelques jours), tout est automatique et on obtient -- avec de la patience -- des fichiers
PDF de résultats avec des graphiques, des fichiers Excel que l'on peut ensuite reprendre et analyser
comme ceux de l'archive des résultats. Remarque : il a fallu environ 16 heures de calcul sur un petit serveur de 2 processeurs 4 coeurs et 32 Go RAM pour réaliser l'analyse de 4 facteurs sur les séquences du run que nous avons déposées à l'ENA.
Utilisé sur des serveurs en parallèle comme ceux de GenScale ou GenOuest, ce script permet d'avoir une démarche commune et des résultats
au même format pour toutes les analyses, à moindre coût humain de manipulation et de saisie.
Ce premier script montre qu'il est possible d'automatiser de nombreux traitements spécifiques à la métagénomique et ouvre la voie à d'autres scripts (pour ITS de façon sure, pour gyrB peut-être) que nous écrirons courant 2015.
Pour les analyses de type WGS, nous aimerions utiliser le framework nommé MetAMOS plutôt que des outils logiciels séparés. En attendant d'installer et de maitriser ce logiciel chronophage dévoreur de ressources informatiques, nous allons utiliser un outil performant
nommé COMMET qui permet d'analyser des reads afin de détecter les reads communs et spécifiques. Via cet outil, nous allons nous focaliser sur les reads spécifiques associés à divers biotopes.
|  Retour à la page principale de
(gH)
Retour à la page principale de
(gH)