![]()
![]()
GREBA 210611 : taille de fenêtre d'analyse ; bioiksemlPartie un : taille de fenêtre d'analyse de séquences
Partie deux : bioikseml
|
 Retour à la page principale de
(gH)
Retour à la page principale de
(gH)


GREBA 210611 : taille de fenêtre d'analyse ; bioikseml
Partie un : taille de fenêtre d'analyse de séquences
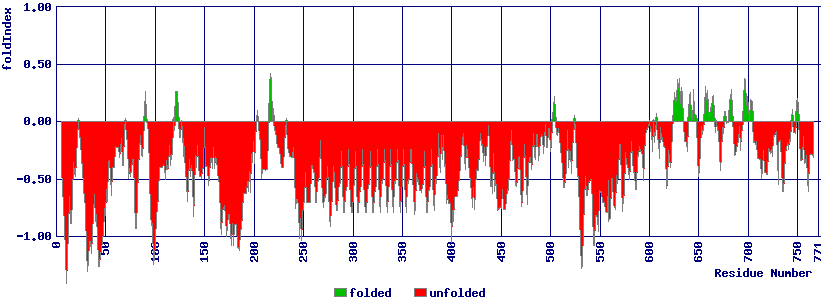
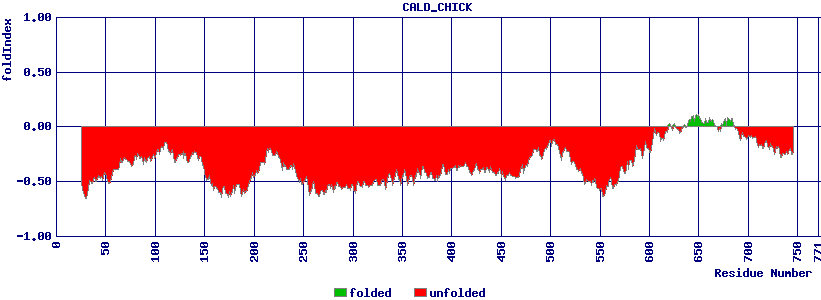
A priori, un seul acide aminé, voire même un seul nucléotide suffit à faire une différence, comme le montre la définition d'un SNP, d'où les sites dbSNP et SNPedia. En ce sens, on pourrait donc dire que la taille minimale en analyse de séquences d'acides aminés est donc de 1/3 puisqu'il faut 3 codons pour faire un acide aminé, ou plutôt de 1/2 si on suit la règle du wobble connue depuis 1996 (Crick). Toutefois, ce n'est pas une valeur particulière qui est intéressante, mais un ensemble de valeurs successives présentant des valeurs similaires (toutes fortes, toutes faibles...) qu'on appréhende selon la technique de calcul nommée moyenne glissante ou mobile. Un exemple de visualisation est fourni par le site du calcul du FoldIndex : sélectionner par exemple CALD_CHICK comme séquence et essayer des fenêtres de taille 10 et 50 :
Il y a deux concepts importants derrière la notion de fenêtre : le premier est celui d'une étude locale de proche en proche (plutôt que partielle) de la séquence, par opposition à un calcul global sur l'ensemble de la séquence. Le second est celui de la prise en compte de de la «séquentialité» des acides aminés. Ainsi MASQQERQQLDARAR n'est pas AAADELMQQQQRRRS (même si tous les calculs effectués sur l'ensemble de la chaine donnent les mêmes valeurs car ce sont les mêmes acides aminés mis par ordre alphabétique) ni QQMASERQQLRARAD (alors qu'il s'agit des mêmes acides aminés mais dans un ordre différent). En gros, local et ordonné versus global et invariant par permutation.
Voici quelques tailles de fenêtres trouvées sur Internet :
Tailles 13 et 17
PHD Profile network from Heidelberg et PROFPHD
Kyte-Doolittle Hydropathy Plots: When looking for surface regions in a globular protein, a window size of 9 was found to give the best results. Surface regions can be identified as peaks below the mid line.When looking for a transmembrane region in a protein, a window size of 19 is needed. Transmembrane regions are identified by peaks with scores greater than 1.6 using a window size of 19.
PONDR (Predictor Of Naturally Disordered Regions)
Selon ieeexplore 4133195: Computational Intelligence and Bioinformatics and Computational Biology, 2006. CIBCB '06. 2006 IEEE Symposium on Chen, Ke Kurgan, Lukasz Ruan, Jishou Dept. of Electr. & Comput. Eng., Alberta Univ., Edmonton, Alta. Optimization of the Sliding Window Size for Protein Structure Prediction Sliding window based methods are relatively often applied in prediction of various aspects related to protein structure. Despite their wide spread use, researchers did not establish a standard related to the size of the window, i.e., window sizes ranging between 7 and 17 residues were used in the past. To this end, this paper performs a computational study based on a probabilistic approach that aims at finding an optimal sliding window size. The results shows that formation of helical structure can be affected by amino acids (AAs) that are up to 9 positions away in the sequence, while the formation of coils and strands can be affected by AAs that are up to 3 and 6 positions away, respectively. Overall, our results suggest that a sliding window with 19 residues is optimal for secondary structure prediction, while for a specific prediction tasks, such as prediction of p-strands, a smaller window size is sufficient. Finally, the 20 AAs are categorized into five groups based on their influence of formation of the secondary structure. The finding related to the optimal window size was confirmed based on an independent experimental study related to the prediction of secondary protein structure.
Selon Pearlman et Wang7 to 10 is optimal pour interior hydrophobic and exterior hydrophylic regions, 6 to 7 is optimal for antigenic regions in Formulation, characterization, and stability of protein drugs 1996 Rodney Pearlman, Y. John Wang.
For protein sequences, use short windows e.g. 1 for window size[...] for dot matrix ; longer window size 15. Bioinformatics: sequence and genome analysis David W. Mount 2004. Window size refers to the number of amino acids examined at a time to determine a point of hydrophobic character. Window size can be varied from 5 to 25 (default 7); one should choose a window that corresponds to the expected size of the structural motif under investigation Window size of 5-7 is good for finding hydrophilic regions that are likely exposed on the surface and may possibly be antigenic. Window size of 19-21 will make hydrophobic, membrane-spanning domains stand out rather clearly (typically > 1.6 on the Kyte-Doolittle scale).
Sliding window-base method to detect mutation rates Fares, Elena. J Mol Evol 2002 55:509-521.
DWih, in protein & peptide Letters, avr. 2000 page 103.
Emboss oddcomp. Find protein sequence regions with a biased composition. Description: oddcomp searches a series of protein files, reporting the identifier for those that exceed a certain amino acid composition threshold in a portion of the sequence. oddcomp was written to answer the question 'which proteins contain at least n X and m Y in p residues'. One could search for serine rich or polyglutamine rich, collagen helix, or similar proteins using this program.
Peter HOJRUP. Secondary structures (databases.ppt)
Partie deux : bioikseml
Le site bioikseml qui se prononce comme BioXML est développé par J. M. RICHER et facilite la conversion de séquences Fasta en fichiers XML. De plus, il permettra à terme de compléter ces fichiers.
Sites sans doute en rapport avec Knowledge Semantics Extraction et bioinformatique :