![]()
![]()
|
Partie Statistiques du cours de BioInformatiqueMaster BTV, UFR Sciences - Université d'AngersSolutions du TD numéro 3 (énoncés)
|
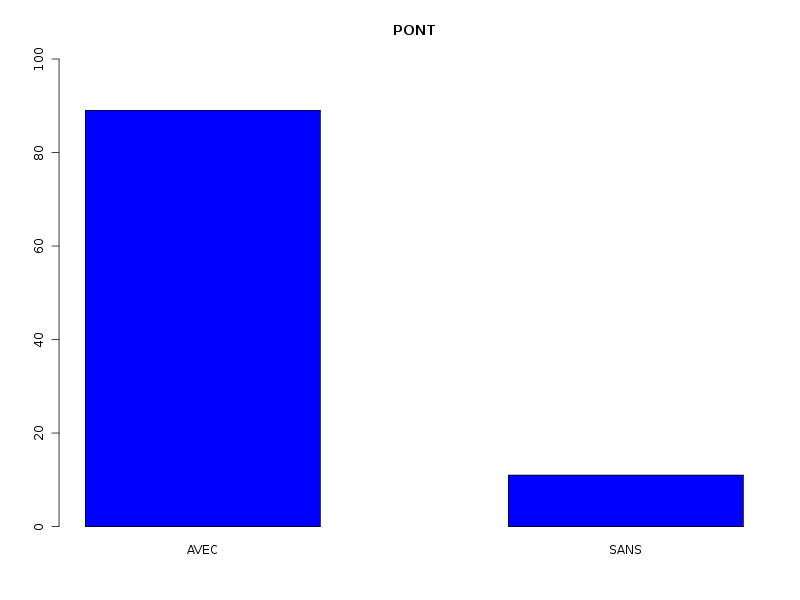
 Retour à la page principale de
(gH)
Retour à la page principale de
(gH)


Partie Statistiques du cours de BioInformatique
Master BTV, UFR Sciences - Université d'Angers
Solutions du TD numéro 3 (énoncés)
Avec nos fonctions, l'étude de toutes les QT peut se faire simplement par
allQT(leaQT,names(leaQT),"aa ? uma ?",TRUE)dont les résultats sont
RESUMES STATISTIQUES Par cdv décroissant Num Nom Taille Moyenne Unite Ecart-type Coef. de var. Minimum Maximum 2 foldindex 773 -0.100 ? 0.151 151.60 % -0.450 0.412 5 gravy 773 -1.061 ? 0.801 75.51 % -9.000 0.799 4 mw 773 21838.965 uma 16041.950 73.46 % -9.000 197129.312 1 length 773 205.688 aa 148.596 72.24 % 68.000 1864.000 3 pi 773 7.377 ? 2.439 33.06 % -9.000 12.610 Par ordre d'entrée Num Nom Taille Moyenne Unite Ecart-type Coef. de var. Minimum Maximum 1 length 773 205.688 aa 148.596 72.24 % 68.000 1864.000 2 foldindex 773 -0.100 ? 0.151 151.60 % -0.450 0.412 3 pi 773 7.377 ? 2.439 33.06 % -9.000 12.610 4 mw 773 21838.965 uma 16041.950 73.46 % -9.000 197129.312 5 gravy 773 -1.061 ? 0.801 75.51 % -9.000 0.799 Par moyenne décroissante Num Nom Taille Moyenne Unite Ecart-type Coef. de var. Minimum Maximum 4 mw 773 21838.965 uma 16041.950 73.46 % -9.000 197129.312 1 length 773 205.688 aa 148.596 72.24 % 68.000 1864.000 3 pi 773 7.377 ? 2.439 33.06 % -9.000 12.610 2 foldindex 773 -0.100 ? 0.151 151.60 % -0.450 0.412 5 gravy 773 -1.061 ? 0.801 75.51 % -9.000 0.799 MATRICE DES COEFFICIENTS DE CORRELATION LINEAIRE length foldindex pi mw gravy length 1.000 foldindex 0.173 1.000 pi -0.195 0.026 1.000 mw 0.992 0.161 -0.151 1.000 gravy 0.119 0.580 0.442 0.191 1.000 Meilleure corrélation 0.9922021 pour mw et length p-value 0 Coefficients de corrélation par ordre décroissant 0.992 p-value 0.0000 pour mw et length 0.580 p-value 0.0000 pour gravy et foldindex 0.442 p-value 0.0000 pour gravy et pi -0.195 p-value 0.0000 pour pi et length 0.191 p-value 0.0000 pour gravy et mw 0.173 p-value 0.0000 pour foldindex et length 0.161 p-value 0.0000 pour mw et foldindex -0.151 p-value 0.0000 pour mw et pi 0.119 p-value 0.0009 pour gravy et length 0.026 p-value 0.4622 pour pi et foldindexOn en conclut que le foldindex varie beaucoup plus que les autres variables, que gravy est "assez homogène" avec un CV proche de ceux de mw et length alors que pi est très peu variable. Compte-tenu du grand nombre de valeurs, presque toutes les corrélations sont considérées comme statistiquement significatives, mais sans doute seules celles entre mw et length, gravy et foldindex, gravy et pi méritent d'être étudiées car supérieures à 0.44, ce qui est élevé ici, pour 773 valeurs...
Comme au TD2, le fichier lea4.dar est une partie du fichier lea.dar : on a enlevé les 3 protéines de règne mineur et celles dont le pi, le mw ou le gravy est -9.
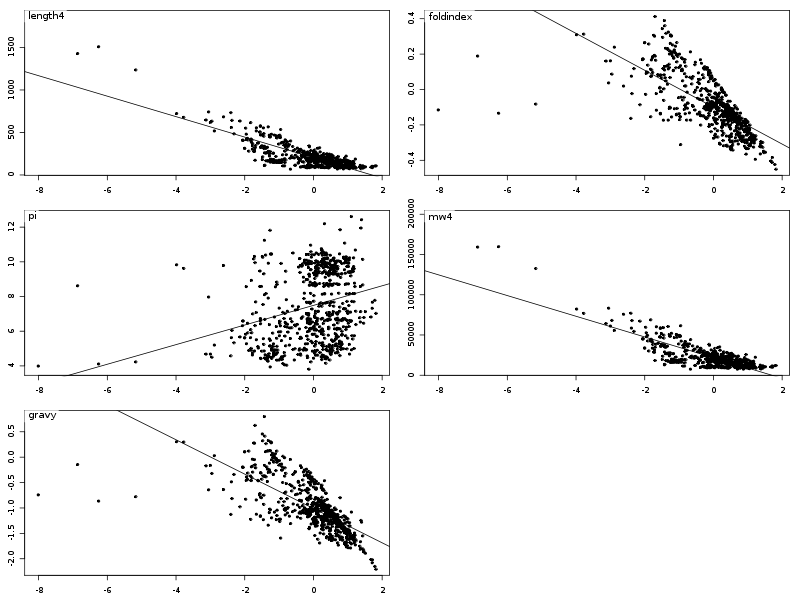
Les résultats s'en ressentent légèrement et en particulier, seules deux corrélations semblent intéressantes, comme le montrent les diagrammes de dispersion obtenus avec la fonction pairs().
RESUMES STATISTIQUES Par cdv décroissant Num Nom Taille Moyenne Unite Ecart-type Coef. de var. Minimum Maximum 2 foldindex 765 -0.100 ? 0.151 150.68 % -0.450 0.412 4 mw4 765 21858.953 uma 15795.787 72.26 % 7521.360 197129.312 1 length4 765 204.797 aa 147.303 71.93 % 68.000 1864.000 5 gravy 765 -1.010 ? 0.483 47.86 % -2.206 0.799 3 pi 765 7.487 ? 2.055 27.45 % 3.810 12.610 Par ordre d'entrée Num Nom Taille Moyenne Unite Ecart-type Coef. de var. Minimum Maximum 1 length4 765 204.797 aa 147.303 71.93 % 68.000 1864.000 2 foldindex 765 -0.100 ? 0.151 150.68 % -0.450 0.412 3 pi 765 7.487 ? 2.055 27.45 % 3.810 12.610 4 mw4 765 21858.953 uma 15795.787 72.26 % 7521.360 197129.312 5 gravy 765 -1.010 ? 0.483 47.86 % -2.206 0.799 Par moyenne décroissante Num Nom Taille Moyenne Unite Ecart-type Coef. de var. Minimum Maximum 4 mw4 765 21858.953 uma 15795.787 72.26 % 7521.360 197129.312 1 length4 765 204.797 aa 147.303 71.93 % 68.000 1864.000 3 pi 765 7.487 ? 2.055 27.45 % 3.810 12.610 2 foldindex 765 -0.100 ? 0.151 150.68 % -0.450 0.412 5 gravy 765 -1.010 ? 0.483 47.86 % -2.206 0.799 MATRICE DES COEFFICIENTS DE CORRELATION LINEAIRE length4 foldindex pi mw4 gravy length4 1.000 foldindex 0.172 1.000 pi -0.249 0.037 1.000 mw4 0.997 0.162 -0.260 1.000 gravy 0.177 0.985 0.017 0.170 1.000 Meilleure corrélation 0.9974722 pour mw4 et length4 p-value 0 Coefficients de corrélation par ordre décroissant 0.997 p-value 0.0000 pour mw4 et length4 0.985 p-value 0.0000 pour gravy et foldindex -0.260 p-value 0.0000 pour mw4 et pi -0.249 p-value 0.0000 pour pi et length4 0.177 p-value 0.0000 pour gravy et length4 0.172 p-value 0.0000 pour foldindex et length4 0.170 p-value 0.0000 pour gravy et mw4 0.162 p-value 0.0000 pour mw4 et foldindex 0.037 p-value 0.3043 pour pi et foldindex 0.017 p-value 0.6429 pour gravy et pi
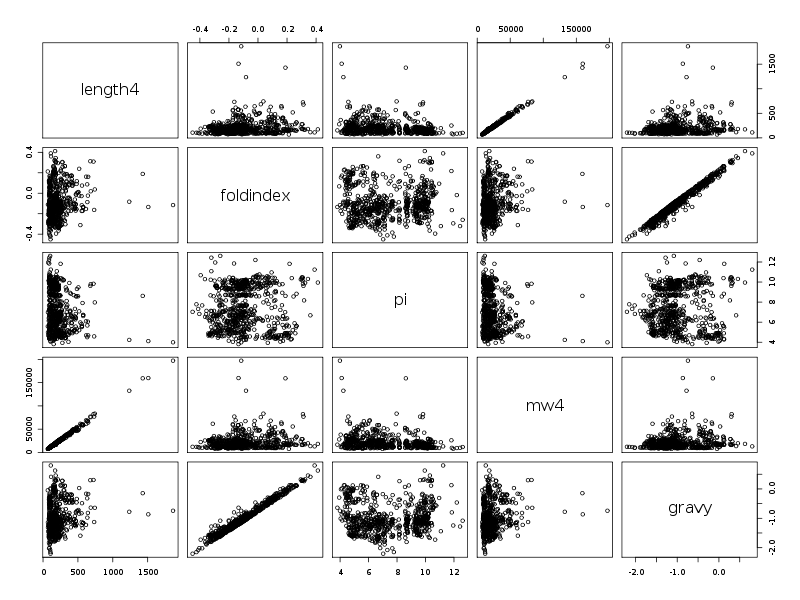
La causalité entre length4 et mw4 est la même que celle du TD2 : plus la longueur augmente, plus la masse moléculaire aussi. Pour essayer de comprendre la liaison entre gravy et foldindex, on peut écrire :
anaLin("Hydropathie moyenne",gravy,"","Indice de repliement",foldindex,"",TRUE)Les résultats et graphiques sont alors perturbés par les données manquantes (valeurs -9) avec le fichier lea.dar :
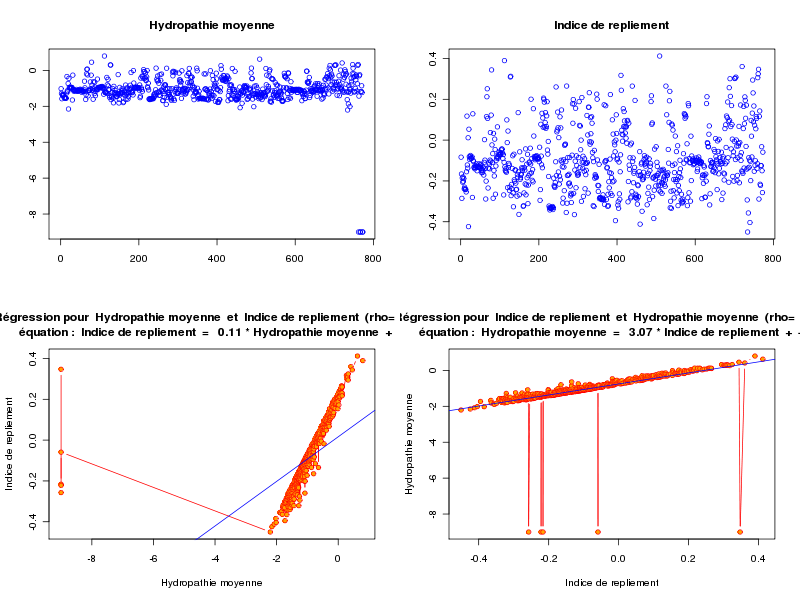
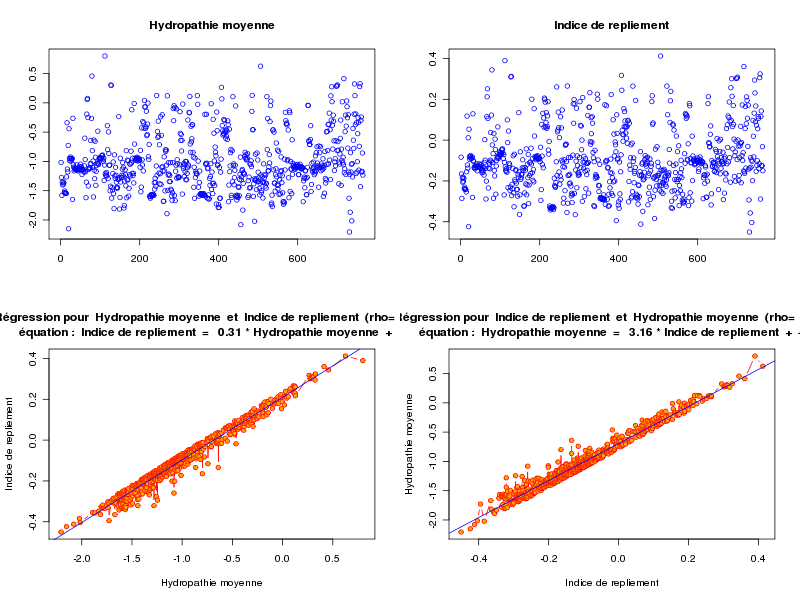
alors qu'avec le fichier lea4.dar on peut "voir des choses" :
ANALYSE DE LA LIAISON LINEAIRE ENTRE Hydropathie moyenne ET Indice de repliement coefficient de corrélation : 0.9854343 donc R2 = 0.9710808 p-value associée : 0 équation : Indice de repliement = 0.31 * Hydropathie moyenne + 0.21 équation : Hydropathie moyenne = 3.16 * Indice de repliement - 0.69
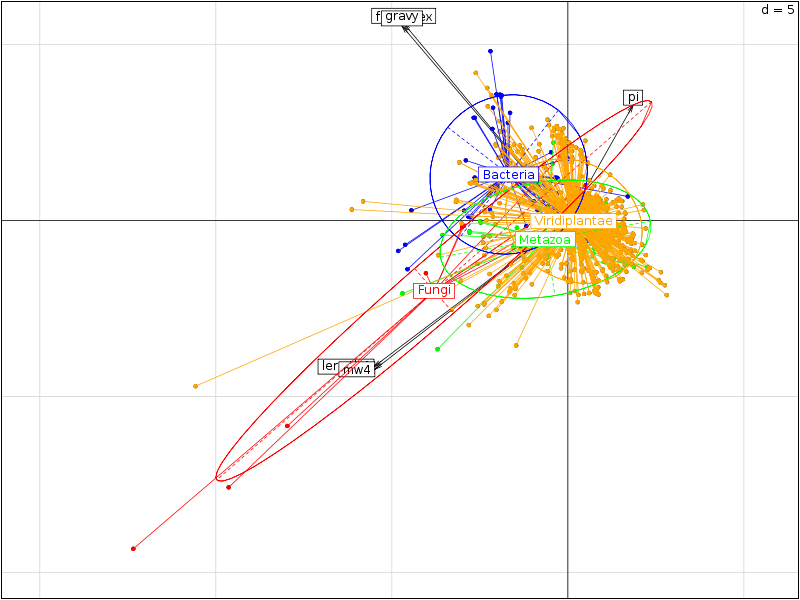
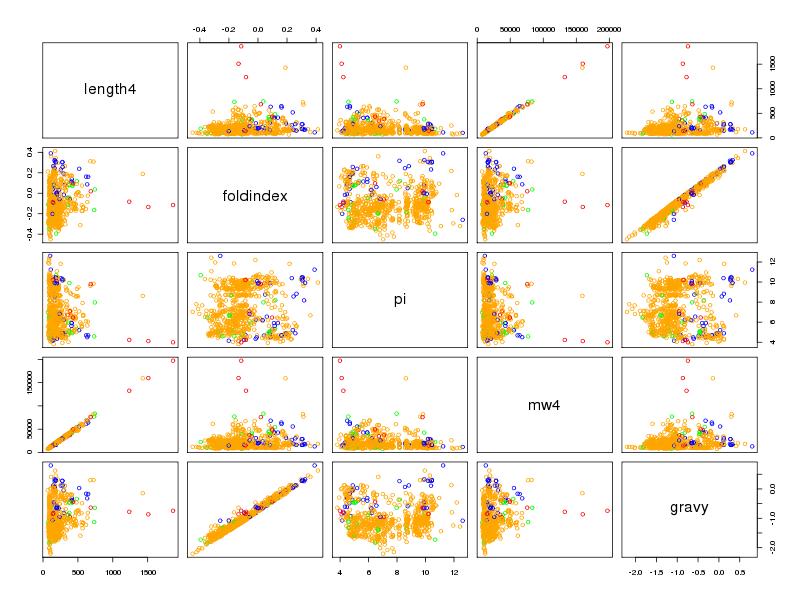
Il est intéressant d'utiliser la couleur pour séparer les différents règnes. On commencera donc par trouver dans la solution ci-dessous des instructions pour définir une couleur pour chaque règne ce qui permet d'obtenir des graphiques de dispersion un petit peu plus lisibles :
Une ACP avec ade4 se fait à l'aide de la fonction dudi.pca ; la composante $eig du résultat contient les valeurs propres que nous avons mises dans la variable vp avant de réaliser un affichage "propre" (sic) de ces valeurs et de les représenter via un histogramme.
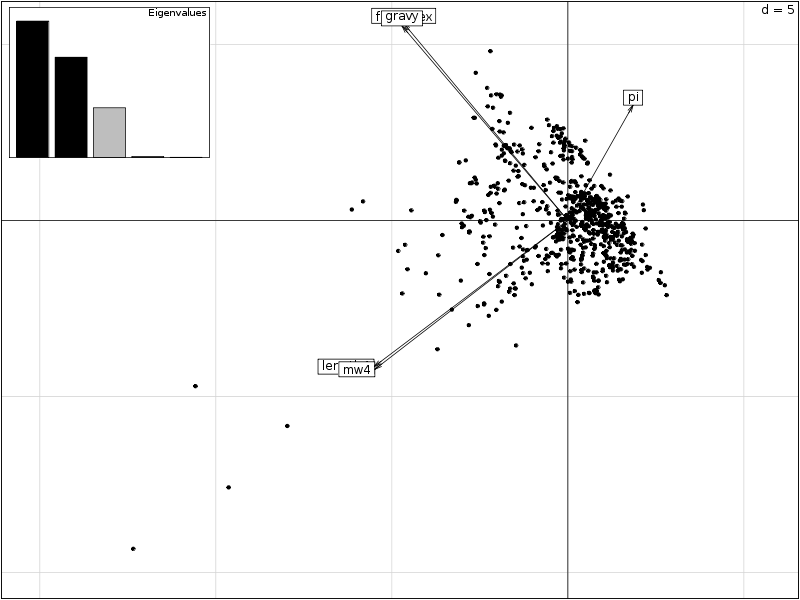
# lecture des données et chargement d'ade4 lea4<-lit.dar("lea4.dar") lea4QT <- lea4[,c(1,5:8)] library(ade4) attach(lea4) # un peu de couleur ne fait pas de mal couleur1 <- rep("blue",length(reign)) couleur1[reign=="Viridiplantae"] <- "orange" couleur1[reign=="Metazoa"] <- "green" couleur1[reign=="Fungi"] <- "red" png("lea_acp1.png",width=800,height=600) pairs(lea4QT,col=couleur1) dev.off() # réalisons l'ACP acp1 <- dudi.pca(lea4QT,scannf = FALSE, nf = 4) # on met en forme les valeurs propres # et on trace son histogramme vp <- acp1$eig vp_i <- 1:length(vp) vp_r <- round( 100*vp/sum(vp),0 ) vp_rc <- cumsum(vp_r) dsc_vp <- cbind(vp_i,vp,vp_r,vp_rc) colnames(dsc_vp) <- c("Axe","Valeur propre","Pourcent","Cumul") print(dsc_vp) barplot(vp_r,col=heat.colors(length(vp_r))) # quelques tracés pour mieux comprendre # comment se situent les variables # et les groupes score(acp1) s.corcircle(acp1$co) couleur <- c("blue","red","green","orange") scatter(acp1,clab.row=0) couleur2 <- c("blue","red","green","orange") scatter(acp1,clab.row=0,posieig="none") s.class(dfxy=acp1$li,fac=reign,col=couleur2,xax=1,yax=2,add.plot=TRUE)Avec 5 variables, il y a au maximum 4 axes. L'examen des valeurs propres montre que 3 axes suffisent mais qu'avec 2 axes on a déjà 82 % d'inertie :
Axe Valeur propre Pourcent Cumul [1,] 1 2.374083416 47 47 [2,] 2 1.744559806 35 82 [3,] 3 0.864575009 17 99 [4,] 4 0.014517334 0 99 [5,] 5 0.002264435 0 99
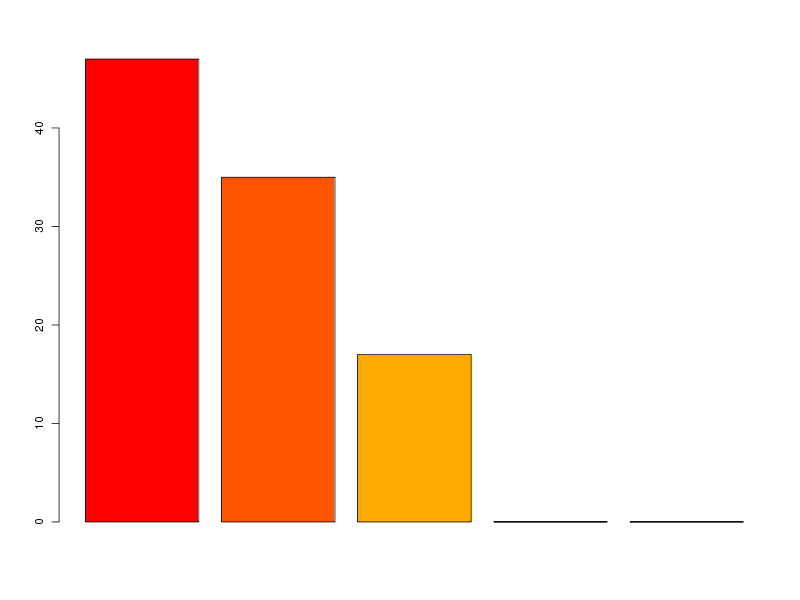
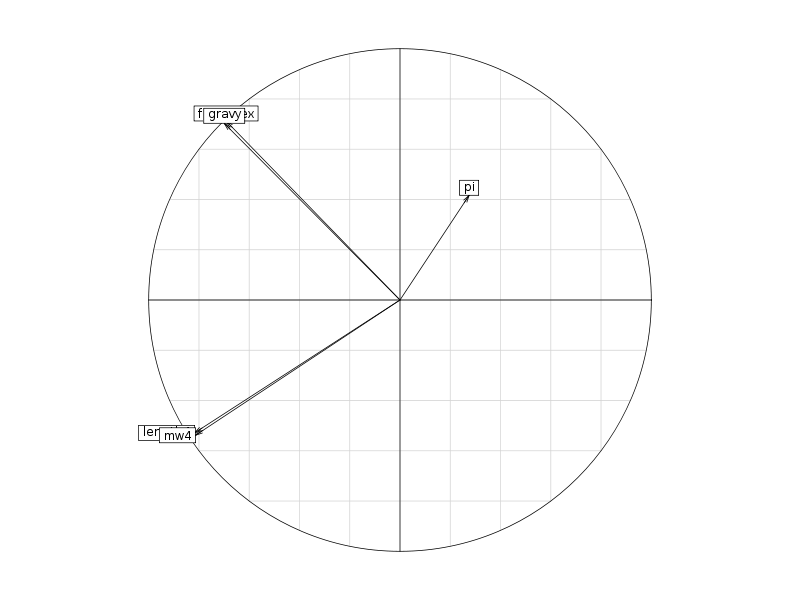
On trouvera sur les graphiques suivant la corrélation des variables avec les axes, le cercle des corrélations, les ellipses de dispersion...
Le cercle des corrélations permet de visualiser ce que nous avions déjà obtenu avec la matrice des corrélations précédente, le graphique avec les ellipses de dispersion confirme ce que montraient les boites à moustaches, à savoir... (conclusion volontairement omise).
Au vu des résultats précédents, on ne devrait conserver que trois variables car les variables length et mw sont équivalentes, de même que gravy et foldindex. Sans justification, nous retiendrons length, gravy et pi. Leur étude par règne, réalisée par
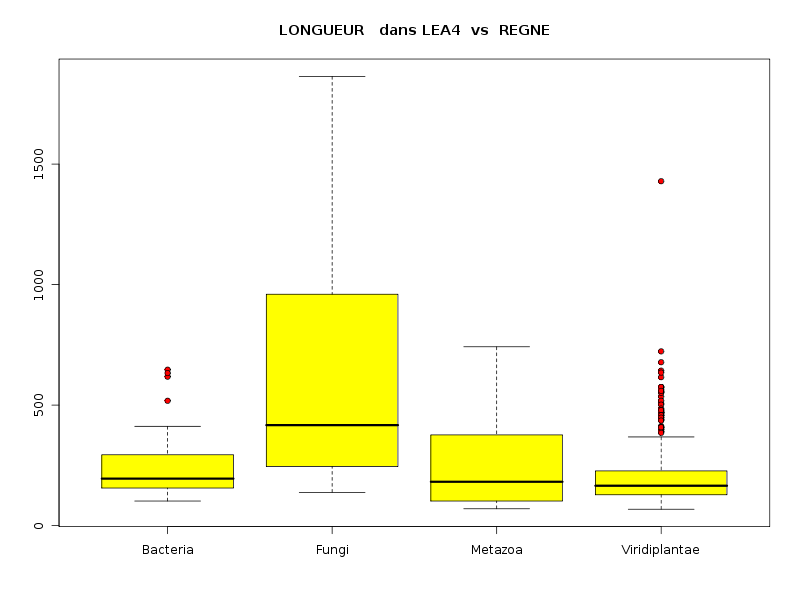
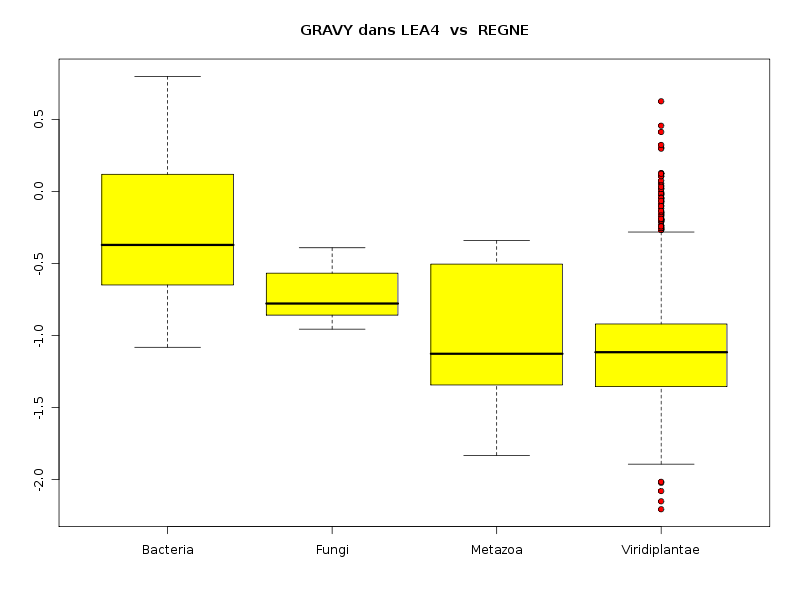
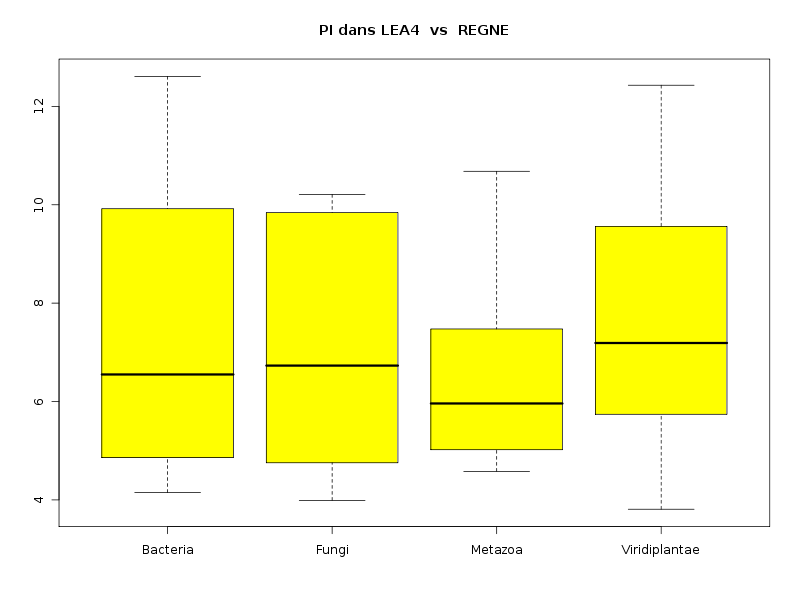
lea4<-lit.dar("lea4.dar") attach(lea4) les4r <- as.character( sort(unique(reign4)) ) decritQTparFacteurTexte("LONGUEUR dans LEA2",length4,"aa","REGNE",reign4,les4r,TRUE,"lgpr1.png") decritQTparFacteurTexte("GRAVY dans LEA2",gravy4,"","REGNE",reign4,les4r,TRUE,"lgpr2.png") decritQTparFacteurTexte("PI dans LEA2",pi4,"","REGNE",reign4,les4r,TRUE,"lgpr3.png") detach(lea4)aboutit aux résultats suivants :
VARIABLE QT LONGUEUR dans LEA2 unité : aa VARIABLE QL REGNE labels : Bacteria Fungi Metazoa Viridiplantae N Moy Unite Ect Cdv Q1 Med Q3 EIQ Min Max Global 765 204.7974 aa 147.3027 72 130 168 234 104 68 1864 Bacteria 37 255.1351 aa 146.2725 57 156 195 294 138 102 647 Fungi 11 663.1818 aa 571.9613 86 245.5 417 960 714.5 138 1864 Metazoa 23 274.4348 aa 193.8123 71 102 182 376.5 274.5 70 742 Viridiplantae 694 192.5403 aa 112.1192 58 128 165.5 227 99 68 1429 Analysis of Variance Response: Df Sum Sq Mean Sq F value Pr(>F) nomVarQL 3 2620832 873611 47.561 < 2.2e-16 *** Residuals 761 13978202 18368 Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1 -------------------------------------------------------------------- VARIABLE QT GRAVY dans LEA2 VARIABLE QL REGNE labels : Bacteria Fungi Metazoa Viridiplantae N Moy Unite Ect Cdv Q1 Med Q3 EIQ Min Max Global 765 -1.0100 0.4834 -48 -1.322 -1.099 -0.8095 0.5126 -2.2056074 0.7990991 Bacteria 37 -0.2840 0.4646 -164 -0.6487 -0.371 0.1193 0.768 -1.0813725 0.7990991 Fungi 11 -0.7167 0.1870 -26 -0.8583 -0.7777 -0.567 0.2913 -0.9555844 -0.3904077 Metazoa 23 -1.0005 0.4513 -45 -1.343 -1.126 -0.5041 0.8389 -1.8317647 -0.3402893 Viridiplantae 694 -1.0537 0.4554 -43 -1.354 -1.116 -0.9202 0.4337 -2.2056074 0.6260116 Analysis of Variance Response: Df Sum Sq Mean Sq F value Pr(>F) nomVarQL 3 21.776 7.259 35.187 < 2.2e-16 *** Residuals 761 156.989 0.206 Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1 -------------------------------------------------------------------- VARIABLE QT PI dans LEA2 VARIABLE QL REGNE labels : Bacteria Fungi Metazoa Viridiplantae N Moy Unite Ect Cdv Q1 Med Q3 EIQ Min Max Global 765 7.4874 2.0553 27 5.62 7.13 9.57 3.95 3.8099999 12.6099997 Bacteria 37 7.3941 2.5712 35 4.86 6.55 9.92 5.06 4.1500001 12.6099997 Fungi 11 7.0900 2.4322 34 4.755 6.73 9.84 5.085 3.99 10.21 Metazoa 23 6.4357 1.7599 27 5.02 5.96 7.475 2.455 4.5799999 10.6800003 Viridiplantae 694 7.5335 2.0164 27 5.74 7.19 9.56 3.82 3.8099999 12.4300003 Analysis of Variance Table Response: nomVarQT Df Sum Sq Mean Sq F value Pr(>F) nomVarQL 3 29.0 9.7 2.2954 0.07651 . Residuals 761 3202.6 4.2 Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Globalement le règne influence donc de façon significative les trois variables, même si l'influence pour pi est nettement moindre que pour length et gravy.
Malheureusement, non. Mysql est un logiciel de base de données. Il existe bien des fonctions de récapitulation par groupe en MySql qui sont : min, max, sum, count, avg, std... mais il n'y a pas de fonction statistique (loi normale, de Student...). De plys MySql travaille avec des "tables" de valeurs et renvoie des lignes et des "tables" de résultats uniquement ; ainsi il n'est pas possible de mettre la moyenne d'un groupe dans une variable. Et MySql ne produit aucun graphique.
Php est un langage de programmation qui n'est pas dédié aux statistiques. Même avec des bibliothèques de de sous-programmes statistiques comme pecl/stats et des blibliothèques graphiques comme jpgraph, php reste un langage peu adapté aux analyses statistiques. On pourra regarder notre page de calculs statistiques en ligne pour s'en rendre compte.
Toutefois il est possible de configurer R en mode "serveur" et s'en servir dans Php. On pourra utiliser le lien Rweb (Lyon) pour le tester avec l'encadré suivant : (qu'on viendra "copier/coller" dans le panneau de saisie).
lea <- read.table("http://forge.info.univ-angers.fr/~gh/Datasets/lea.dar", head=TRUE,row.names=1) source("http://forge.info.univ-angers.fr/~gh/statgh.r") lng <- lea[,1] decritQT("Longueur des protéines de LEAPDB",lng,"aa",TRUE)L'intérêt évident est qu'on peut alors faire "dialoguer" R et php, le premier pour le calcul et le second pour les pages Web.
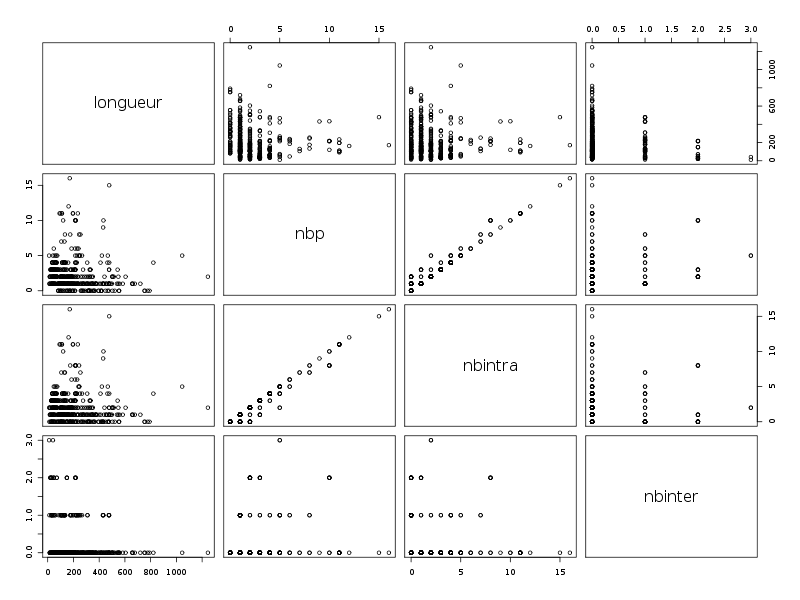
La requête MySql extrait les champs que l'on trouve dans le fichier dbdb.dar. Les données proviennent de deux tables : dbdbprot (dénommée aussi "a") et dbdbchain (dénommée aussi "b"). Afin d'obtenir les résultats sur les ponts et sur la longueur des chaines, on joint les deux tables par la condition a.pr_id = b.ch_pr_id. Le premier champ extrait est length, (renommé "longueur") qui donne la longueur de la chaine ; le second champ, renommé "nbp" donne le nombre total de ponts (intra plus inter) ; les troisièmes et quatrièmes champs sont simplement le nombre de ponts intra et le nombre de ponts inter renommés respectivement "nbintra" et "nbinter" ; le cinquième champ est une indication de présence d'un ou plusieurs ponts, indifféremment inter ou intra. Quand il y a au moins un pont, ce champ nommé "pont" a par valeur "AVEC" ; sinon, il contient "SANS". Enfin, le sixième et dernier champ donne la nature des ponts éventuellement présents. Ses valeurs sont "AUCUN", "INTER_SEUL", "INTRA_SEUL" et "INTER_ET_INTRA" ; ce champ est nommé "naturePont".
Clairement les champs longueur, nbp, nbintra et inter sont des variables QT alors que les champs pont et naturePont sont des variables QL. En voici une étude à une dimension :
# chargement des fonctions gH source("statgh.r") # lecture des données dbdb <- lit.dar("dbdb.dar") attach(dbdb) # séparation des Qt et des QL dbdbQT <- dbdb[,(1:4)] dbdbQL <- dbdb[,(5:6)] # analyse des QT allQT(dbdbQT,names(dbdbQT),"aa ponts ponts ponts",TRUE) # analyse des QL decritQL("PONT",pont,levels(pont),TRUE) decritQL("NATUREPONT",naturePont,levels(naturePont),TRUE) triCroise("PONT",pont,levels(pont), "NATUREPONT",naturePont,levels(naturePont),TRUE) detach(dbdb)Les résultats numériques et graphiques sont :
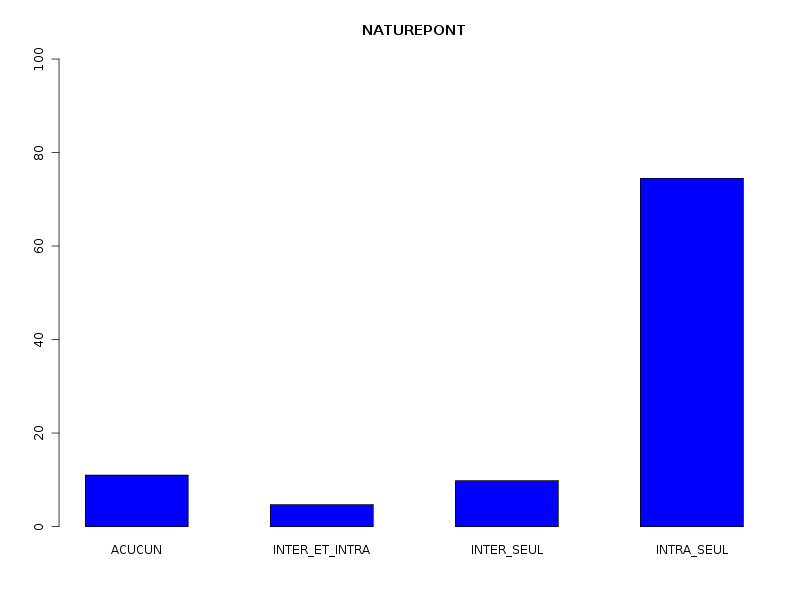
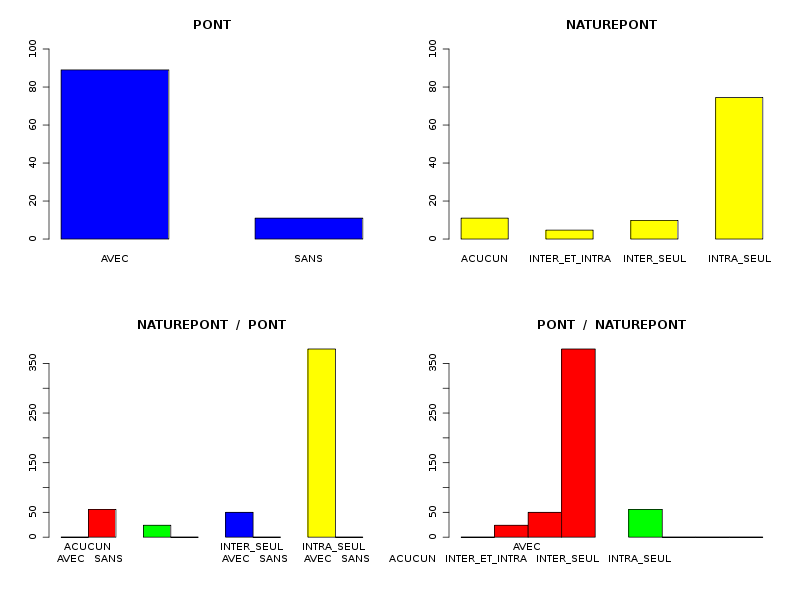
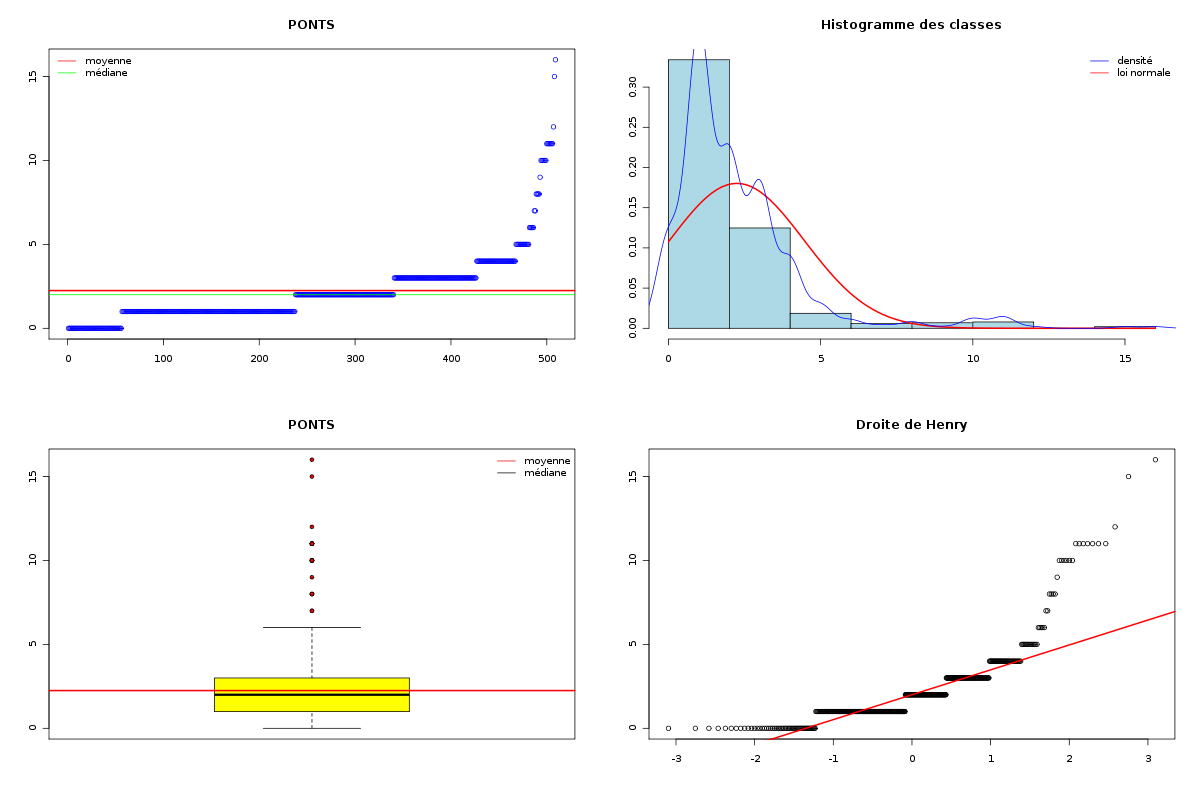
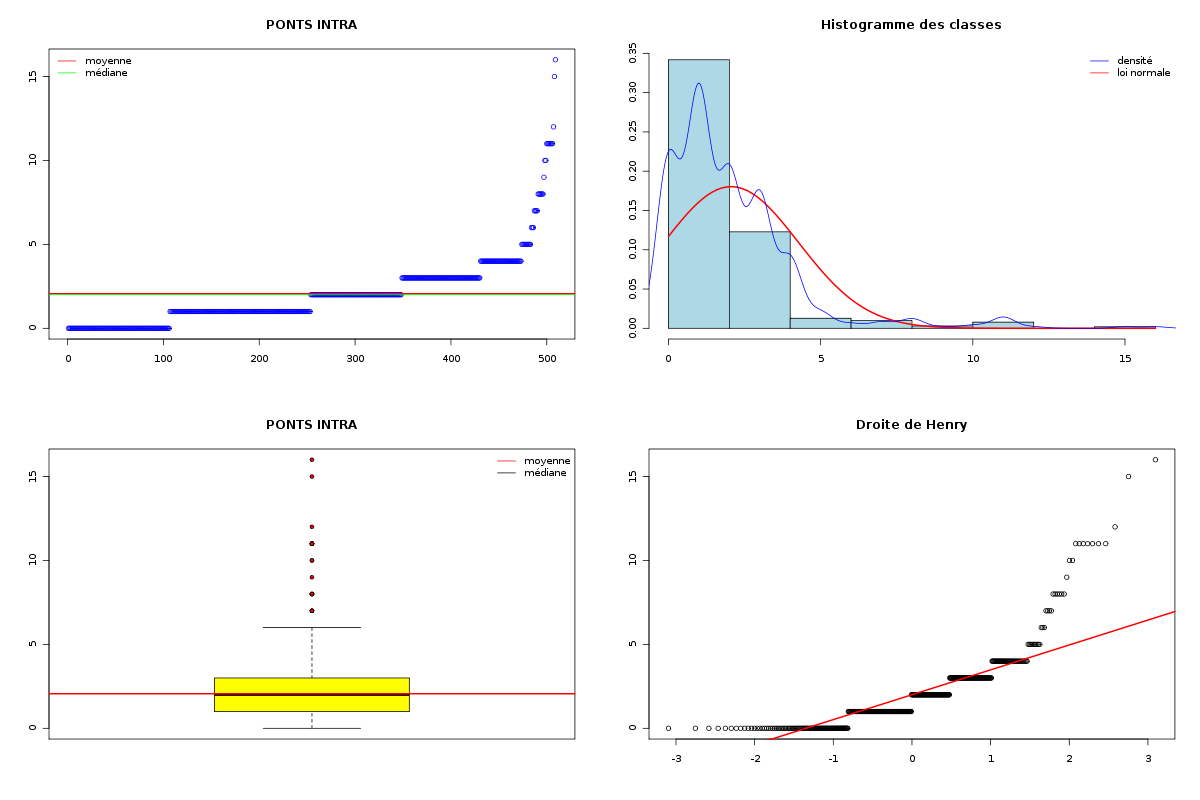
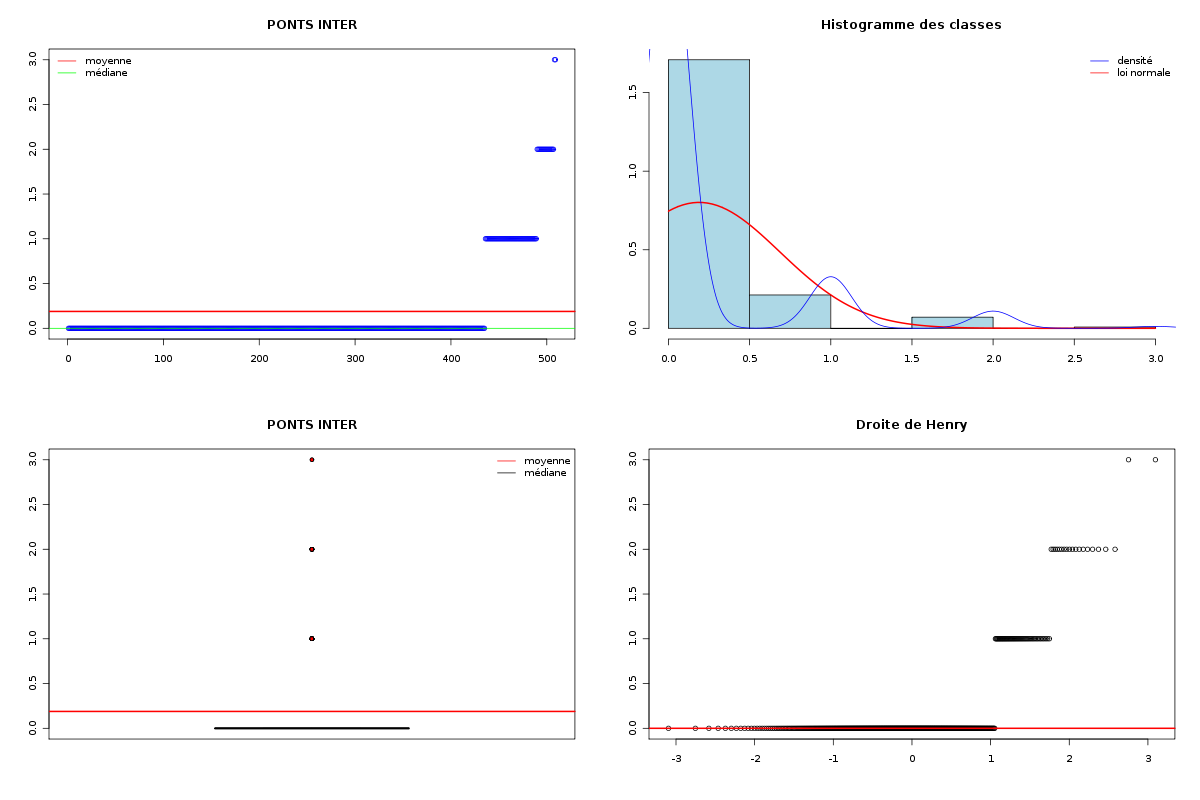
Par cdv décroissant Num Nom Taille Moyenne Unite Ecart-type Coef. de var. Minimum Maximum 4 nbinter 509 0.189 ponts 0.497 263.69 % 0.000 3.000 3 nbintra 509 2.059 ponts 2.210 107.34 % 0.000 16.000 2 nbp 509 2.248 ponts 2.211 98.38 % 0.000 16.000 1 longueur 509 191.778 aa 165.375 86.23 % 11.000 1245.000 Matrice des corrélations longueur nbp nbintra nbinter longueur 1.000 nbp -0.123 1.000 nbintra -0.099 0.975 1.000 nbinter -0.109 0.115 -0.110 1.000 Meilleure corrélation 0.974695 pour nbintra et nbp p-value 0 Formules nbp = 0.975 * nbintra + 0.240 et nbintra = 0.974 * nbp - 0.131 Coefficients de corrélation par ordre décroissant 0.975 p-value 0.0000 pour nbintra et nbp -0.123 p-value 0.0055 pour nbp et longueur 0.115 p-value 0.0096 pour nbinter et nbp -0.110 p-value 0.0128 pour nbinter et nbintra -0.109 p-value 0.0139 pour nbinter et longueur -0.099 p-value 0.0263 pour nbintra et longueur QUESTION : PONT (ordre des modalités) AVEC SANS Total Effectif 453 56 509 Frequence (en %) 89 11 100 QUESTION : PONT (par fréquence décroissante) AVEC SANS Total Effectif 453 56 509 Frequence (en %) 89 11 100 QUESTION : NATUREPONT (ordre des modalités) ACUCUN INTER_ET_INTRA INTER_SEUL INTRA_SEUL Total Effectif 56 24 50 379 509 Frequence (en %) 11 5 10 74 100 QUESTION : NATUREPONT (par fréquence décroissante) INTRA_SEUL ACUCUN INTER_SEUL INTER_ET_INTRA Total Effectif 379 56 50 24 509 Frequence (en %) 74 11 10 5 100 TRI CROISE DES QUESTIONS : PONT (en ligne) NATUREPONT (en colonne) Effectifs ACUCUN INTER_ET_INTRA INTER_SEUL INTRA_SEUL AVEC 0 24 50 379 SANS 56 0 0 0 Valeurs en % du total ACUCUN INTER_ET_INTRA INTER_SEUL INTRA_SEUL TOTAL AVEC 0 5 10 74 89 SANS 11 0 0 0 11 TOTAL 11 5 10 74 100 CALCUL DU CHI-DEUX D'INDEPENDANCE ================================= TABLEAU DES DONNEES AVEC SANS Total ACUCUN 0 56 56 INTER_ET_INTRA 24 0 24 INTER_SEUL 50 0 50 INTRA_SEUL 379 0 379 Total 453 56 509 VALEURS ATTENDUES et MARGES AVEC SANS Total ACUCUN 50 6.2 56 INTER_ET_INTRA 21 2.6 24 INTER_SEUL 44 5.5 50 INTRA_SEUL 337 41.7 379 Total 453 56.0 509 CONTRIBUTIONS SIGNEES AVEC SANS ACUCUN - 49.839 + 403.161 INTER_ET_INTRA + 0.326 - 2.640 INTER_SEUL + 0.680 - 5.501 INTRA_SEUL + 5.155 - 41.697 Valeur du chi-deux 509 Le chi-deux max (table) à 5 % est 7.814728 ; p-value 5.348156e-110 pour 3 degrés de liberte décision : au seuil de 5 % on peut rejeter l'hypothèse qu'il y a indépendance entre ces deux variables qualitatives. PLUS FORTES CONTRIBUTIONS AVEC SIGNE DE DIFFERENCE Signe Valeur Pct Mligne Mcolonne Ligne Colonne Obs Th + 403.161 79.21 % ACUCUN SANS 1 2 56 6.2 - 49.839 9.79 % ACUCUN AVEC 1 1 0 49.8 - 41.697 8.19 % INTRA_SEUL SANS 4 2 0 41.7 - 5.501 1.08 % INTER_SEUL SANS 3 2 0 5.5 + 5.155 1.01 % INTRA_SEUL AVEC 4 1 379 337.3 - 2.640 0.52 % INTER_ET_INTRA SANS 2 2 0 2.6 + 0.680 0.13 % INTER_SEUL AVEC 3 1 50 44.5 + 0.326 0.06 % INTER_ET_INTRA AVEC 2 1 24 21.4
auxquel on ajoutera les graphiques par variable QT
obtenus par les instructions R suivantes :
# chargement des fonctions gH source("statgh.r") # lecture des données dbdb <- lit.dar("dbdb.dar") attach(dbdb) # tracé des QT decritQT("LONGUEUR",longueur,"aa",TRUE,"dbdbana1_5.png") decritQT("PONTS",nbp,"",TRUE,"dbdbana1_6.png") decritQT("PONTS INTRA",nbintra,"",TRUE,"dbdbana1_7.png") decritQT("PONTS INTER",nbinter,"",TRUE,"dbdbana1_8.png") detach(dbdb)Dans la mesure où les nombres de ponts sont des variables QT "discrètes", on peut considérer les nombres de ponts comme des modalités, d'où les instructions :
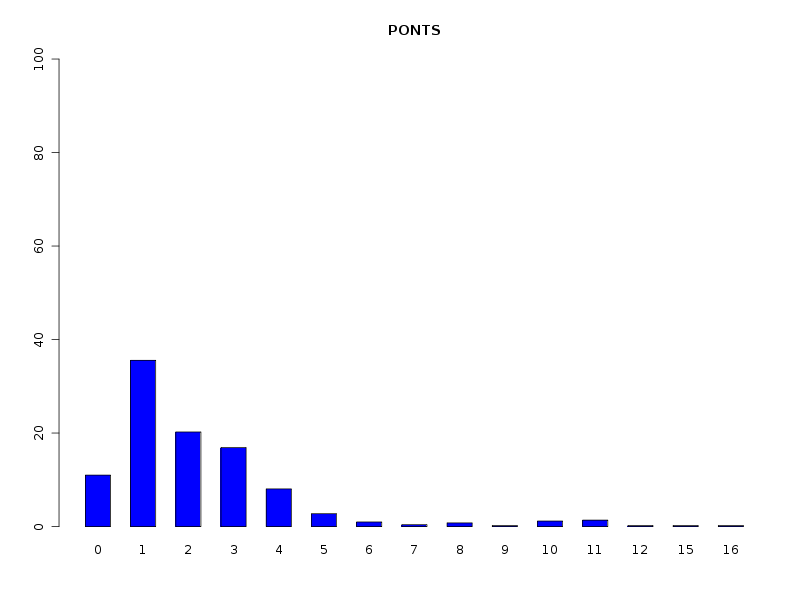
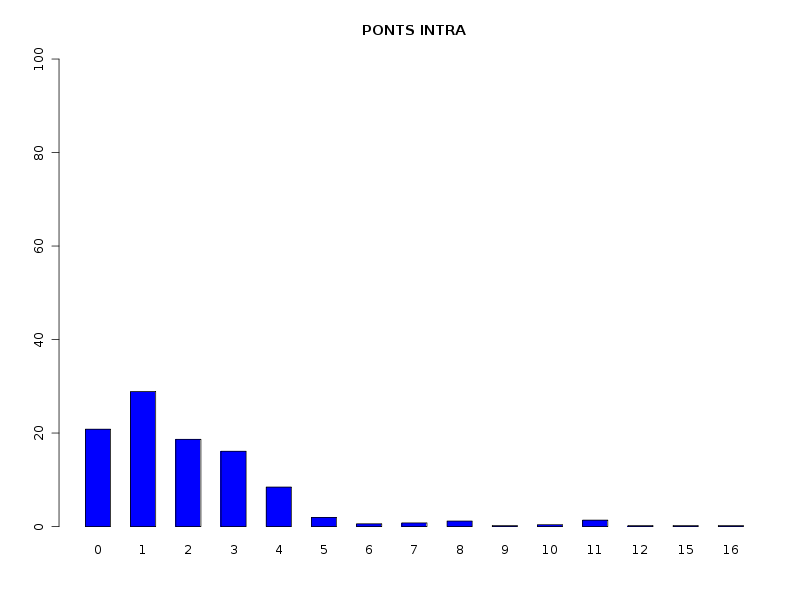
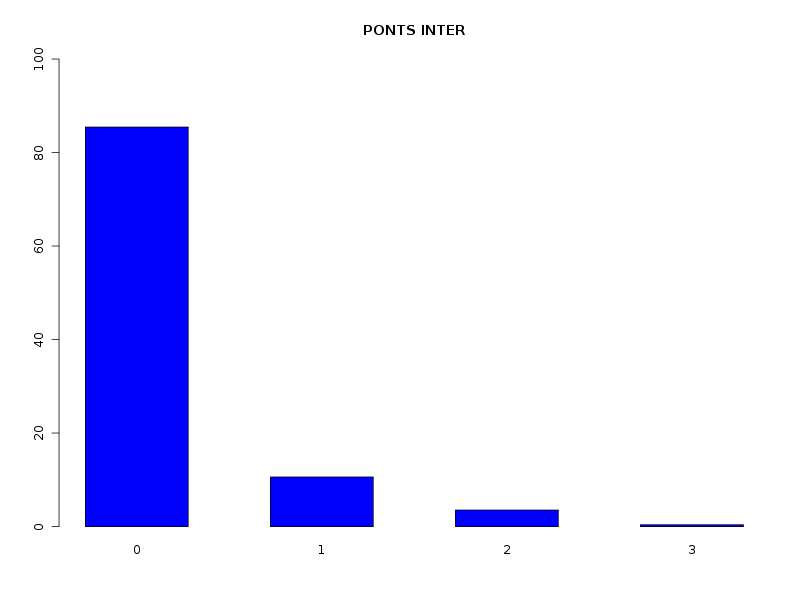
# analyse des QT discrètes attach(dbdb) decritQL("PONTS",nbp,as.character(sort(unique(nbp))),TRUE,"dbdbana1_9.png") decritQL("PONTS INTRA",nbintra,as.character(sort(unique(nbintra))),TRUE,"dbdbana1_10.png") decritQL("PONTS INTER",nbinter,as.character(sort(unique(nbinter))),TRUE,"dbdbana1_11.png") detach(dbdb)En voici les résultats.
QUESTION : PONTS (ordre des modalités) 0 1 2 3 4 5 6 7 8 9 10 11 12 15 16 Total Effectif 56 181 103 86 41 14 5 2 4 1 6 7 1 1 1 509 Frequence (en %) 11 36 20 17 8 3 1 0 1 0 1 1 0 0 0 99 QUESTION : PONTS (par fréquence décroissante) 1 2 3 0 4 5 11 10 6 8 7 9 12 15 16 Total Effectif 181 103 86 56 41 14 7 6 5 4 2 1 1 1 1 509 Frequence (en %) 36 20 17 11 8 3 1 1 1 1 0 0 0 0 0 99 -------------------------------------------------------------------- QUESTION : PONTS INTRA (ordre des modalités) 0 1 2 3 4 5 6 7 8 9 10 11 12 15 16 Total Effectif 106 147 95 82 43 10 3 4 6 1 2 7 1 1 1 509 Frequence (en %) 21 29 19 16 8 2 1 1 1 0 0 1 0 0 0 99 QUESTION : PONTS INTRA (par fréquence décroissante) 1 0 2 3 4 5 11 8 7 6 10 9 12 15 16 Total Effectif 147 106 95 82 43 10 7 6 4 3 2 1 1 1 1 509 Frequence (en %) 29 21 19 16 8 2 1 1 1 1 0 0 0 0 0 99 -------------------------------------------------------------------- QUESTION : PONTS INTER (ordre des modalités) 0 1 2 3 Total Effectif 435 54 18 2 509 Frequence (en %) 85 11 4 0 100 QUESTION : PONTS INTER (par fréquence décroissante) 0 1 2 3 Total Effectif 435 54 18 2 509 Frequence (en %) 85 11 4 0 100
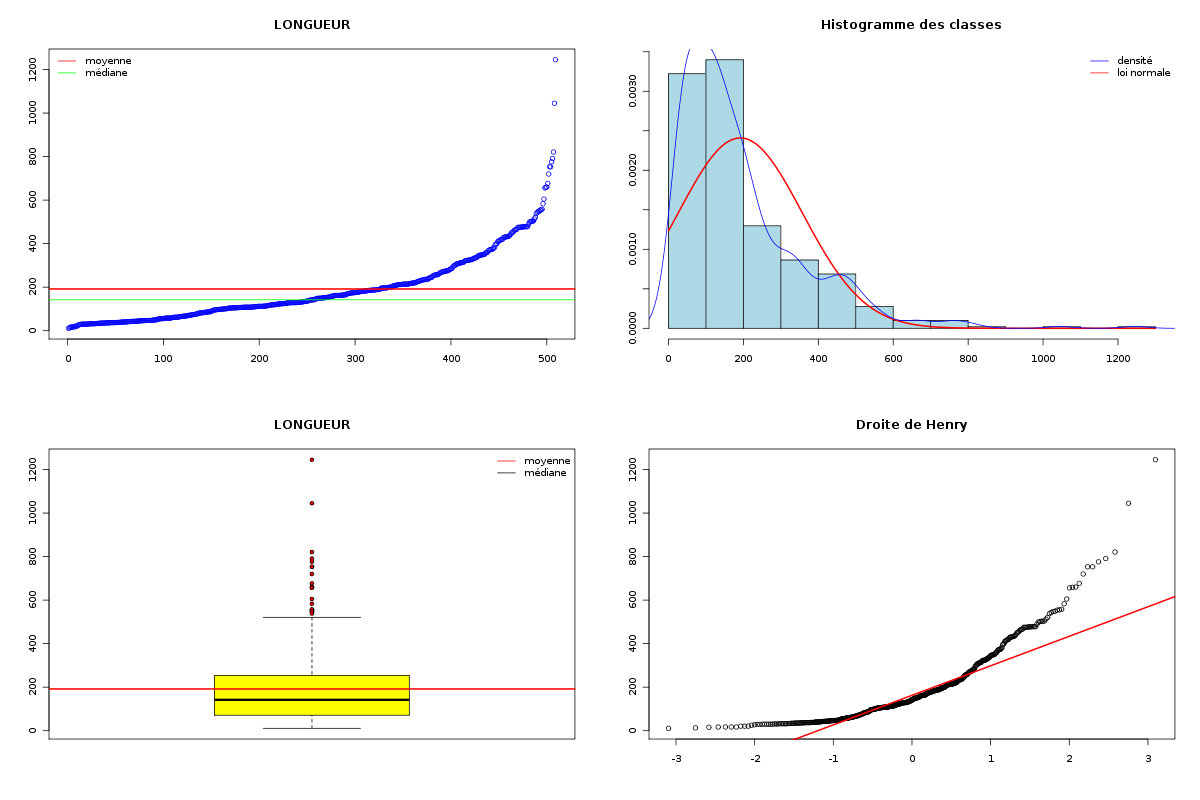
Passons maintenant à la partie rédaction pour ces résultats QT. Les variables nbintra et nbinter sont très dispersées, beaucoup plus que les variables length et ponts, ce qui est un peu surprenant, car après tout nbp n'est autre que la somme de nbintra et de nbinter. La moyenne des longueurs des chaines (car de nombreuses protéines ont plusieurs chaines, notamment pour former des ponts inter) est d'environ 192 aa, avec un cdv de 86 %. On notera qu'il y a une chaine extraordinairement petite, de 11 aa, ce qui indique peut-etre une erreur de saisie (ou d'extraction automatique ?). Le nombre moyen de ponts semble être d'à peu près 2,2 avec 2,1 ponts intra et 0,2 ponts inter en moyenne.
Il ne semble pas y avoir de liaison linéaire entre la longueur et les autres variables ; par contre le nombre de ponts en tout et le nombre de ponts intra sont liés par une relation qui indique qu'à un pont en général de plus, il y a 0.98 pont intra en plus.
La lecture des résultats QL montre que pratiquement 90 % des chaines ont des ponts, et que ces ponts sont en très grande majorité des ponts intra. Une façon plus "propre" d'afficher les pourcentages de ponts avec leur nature serait d'ailleurs plutôt celle-ci :
Sans pont 11 % Avec pont 89 % dont pont intra seul 81 % dont pont inter seul 5 % dont intra et inter 2 %On peut aussi ajouter qu'il y a très peu globalement (5 %) de protéines avec simultanément des ponts INTER et INTRA, pourcentage qui vaut 2 % si on restreint aux protéines avec pont.
Il n'y a pas besoin de lire les tableaux liés au tri croisé : avec "AUCUN" pont, on ne peut pas avoir de pont, donc les modalités "INTER_ET_INTRA", "INTER_SEUL" et "INTRA_SEUL" sont forcément liées à la modalité "AVEC".
Le traitement des QT comme des QT discrètes permet de voir que la majorité des chaines ont peu de ponts (ce qu'on savait déjà). Il serait d'ailleurs prudent d'ajouter les fréquences croissantes via une nouvelle fonction nommée freqCroiss :
######################################## freqCroiss <- function(v) { ######################################## frqa <- table(v) frqr <- table(v)/length(v) frqra <- round(100*frqr) frqrc <- round(100*cumsum(frqr)) frq <- as.data.frame(rbind(frqa,frqra,frqrc)) row.names(frq) <- c("Effectifs ","Pourcentages","Cumul pct") print( frq ) } # fin de fonction freqCroiss ######################################## dbdb <- lit.dar("dbdb.dar") attach(dbdb) freqCroiss( nbp ) detach(dbdb)Il serait également "intelligent" de travailler avec uniquement les protéines à pont, soit les instructions :
cat("Fréquences et fréquences cumulées pour toutes les protéines \n") freqCroiss( nbp ) cat("Fréquences et fréquences cumulées pour les protéines à pont seulement \n") vrai_nbp <- nbp[nbp>0] freqCroiss(vrai_nbp)On voit alors que 90 % des chaines ont au plus 4 ponts, avec 40 % de chaines avec 1 seul pont :
Fréquences et fréquences cumulées pour toutes les protéines 0 1 2 3 4 5 6 7 8 9 10 11 12 15 16 Effectifs 56 181 103 86 41 14 5 2 4 1 6 7 1 1 1 Pourcentages 11 36 20 17 8 3 1 0 1 0 1 1 0 0 0 Cumul pct 11 47 67 84 92 94 95 96 97 97 98 99 100 100 100 Fréquences et fréquences cumulées pour les protéines à pont seulement 1 2 3 4 5 6 7 8 9 10 11 12 15 16 Effectifs 181 103 86 41 14 5 2 4 1 6 7 1 1 1 Pourcentages 40 23 19 9 3 1 0 1 0 1 2 0 0 0 Cumul pct 40 63 82 91 94 95 95 96 96 98 99 100 100 100Il serait tentant de comparer les longueurs en moyenne avec notre fonction compMoyData et d'en déduire que les moyennes ne sont pas significativement différentes via
compMoyData("Longueurs","DBDB",lngDBDB,"LEAPDB",lngLEAPDB)qui fournit comme résultats :
COMPARAISON DE MOYENNES (valeurs fournies) : Longueurs Variable NbVal Moyenne Variance Ecart-type Cdv DBDB 509 191.778 27402.642 165.537 86 % LEAPDB 765 204.797 21726.484 147.399 72 % Global 1274 199.596 24015.226 154.968 78 % Différence réduite : 1.4357 ; p-value 0.1514063 Au seuil de 5 % soit 1.96, on ne peut pas rejeter l'hypothèse d'égalité des moyennes. En d'autres termes, il n'y a pas de différence significative entre les moyennes au seuil de 5 %. Voici ce qu'affiche la fonction t.test de R : Welch Two Sample t-test data: varA and varB t = -1.4357, df = 1000.26, p-value = 0.1514 alternative hypothesis: true difference in means is not equal to 0 95 percent confidence interval: -30.814766 4.775987 sample estimates: mean of x mean of y 191.7780 204.7974 A l'aide d'anova, on obtient Analysis of Variance Table Response: valeurs Df Sum Sq Mean Sq F value Pr(>F) groupes 1 51807 51807 2.1592 0.1420 Residuals 1272 30519576 23993Mais ce serait conclure un peu vite car il y a trois raisons de ne pas faire ce test :
# la longueur dans DBDB ne respecte pas la condition de normalité print( shapiro.test(lngDBDB) ) lea4 <- lit.dar("lea4.dar") attach(lea4) lngLEAPDB <- length4 detach(lea4) # la longueur dans LEAPDB ne respecte pas la condition de normalité print( shapiro.test(lngLEAPDB) ) # il n'y a pas égalité des variances print( var.test(lngDBDB,lngLEAPDB) ) # donc au lieu du test t de Student (paramétrique), on utilise # le test de Wilcoxon -- ou Wilcox -- qui est non paramétrique print( wilcox.test(lngDBDB,lngLEAPDB) )Finalement, on peut conclure que les longueurs sont significativement différentes :
Shapiro-Wilk normality test data: lngDBDB ; W = 0.8311, p-value < 2.2e-16 Shapiro-Wilk normality test data: lngLEAPDB ; W = 0.6313, p-value < 2.2e-16 F test to compare two variances data: lngDBDB and lngLEAPDB F = 1.2613, num df = 508, denom df = 764, p-value = 0.003847 alternative hypothesis: true ratio of variances is not equal to 1 95 percent confidence interval: 1.077409 1.480276 sample estimates: ratio of variances 1.261255 Wilcoxon rank sum test with continuity correction data: lngDBDB and lngLEAPDB W = 162795, p-value = 7.082e-07 alternative hypothesis: true location shift is not equal to 0Voici une autre façon de faire pour les calculs (afin d'afficher les encoches des boites à moustaches, rajouter notch=TRUE dans l'instruction boxplot) :
# on crée un vecteur base qui contient # "DBDB" n1 fois puis "LEAPDB" n2 fois # où n1 est le nombre de chaines dans la DBDB # et n2 est le nombre de chaines dans la LEAPDB n1 <- length(lngDBDB) n2 <- length(lngLEAPDB) base <- c( rep("DBDB",n1), rep("LEAPDB",n2) ) lngs <- c( lngDBDB, lngLEAPDB) decritQTparFacteurTexte("Longueur ",lngs,"aa", "Base",base,"DBDB LEAPDB",TRUE,"complng.png")VARIABLE QT Longueur unité : aa VARIABLE QL Base labels : DBDB LEAPDB N Moy Unite Ect Cdv Q1 Med Q3 EIQ Min Max Global 1274 199.5958 aa 154.9076 78 108 164 238 130 11 1864 DBDB 509 191.7780 aa 165.3747 86 71 142 254 183 11 1245 LEAPDB 765 204.7974 aa 147.3027 72 130 168 234 104 68 1864 Analysis of Variance Table Response: nomVarQT Df Sum Sq Mean Sq F value Pr(>F) nomVarQL 1 51807 51807 2.1592 0.1420 Residuals 1272 30519576 23993
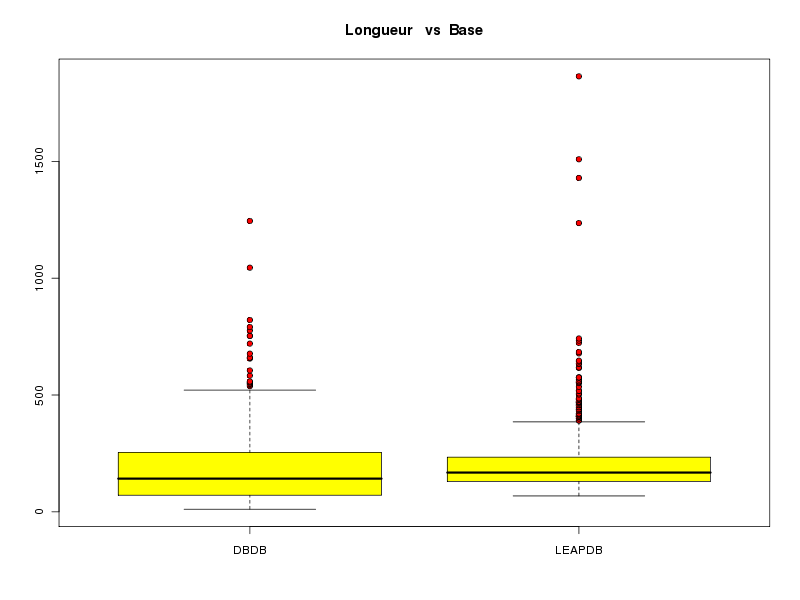
Pour les intervalles de confiance, on peut soit utiliser t.test ou notre fonction icmQT mais là encore, les résultats affichés ne devraient pas être utilisés car l'hypothèse de normalité n'est pas respectée.
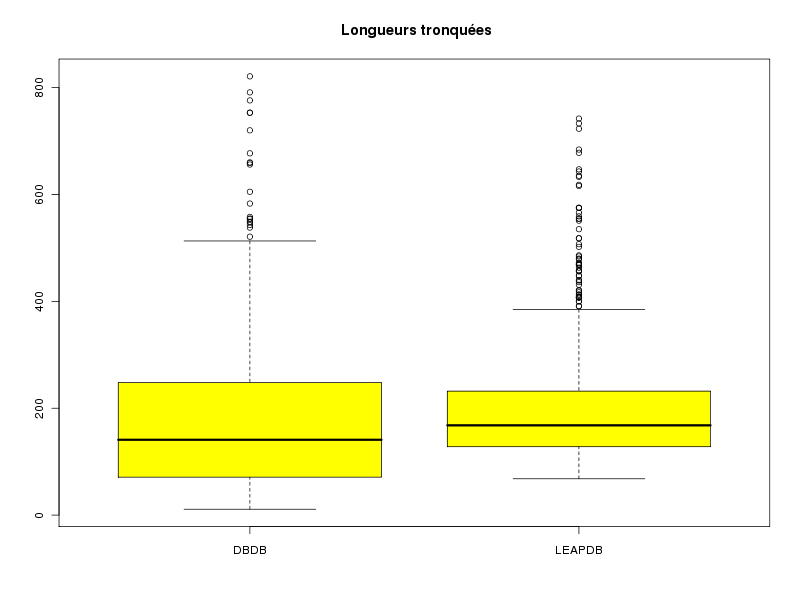
# l'intervalle de confiance est fourni par t.test print(t.test( lngDBDB )) print(t.test( lngLEAPDB )) # ou par notre fonction icmQT cat("pour lngDBDB\n"); icmQT( lngDBDB,0.05,TRUE ) cat("pour lngLEAPDB\n"); icmQT( lngLEAPDB,0.05,TRUE )One Sample t-test data: lngDBDB t = 26.1373, df = 508, p-value < 2.2e-16 alternative hypothesis: true mean is not equal to 0 95 percent confidence interval: 177.3628 206.1932 sample estimates: mean of x 191.778 One Sample t-test data: lngLEAPDB t = 38.4291, df = 764, p-value < 2.2e-16 alternative hypothesis: true mean is not equal to 0 95 percent confidence interval: 194.3357 215.2590 sample estimates: mean of x 204.7974 pour lngDBDB Pour 509 valeurs de moyenne 191.778 et d'écart-type 165.5374 au niveau 0.05 la valeur critique issue de la loi du t de Student pour 508 ddl est 1.964645 soit 1.965 l'intervalle de confiance est donc sans doute [ 177.3628 ; 206.1932 ] soit, en arrondi, [ 177.363 ; 206.193 ] pour lngLEAPDB Pour 765 valeurs de moyenne 204.7974 et d'écart-type 147.3991 au niveau 0.05 la valeur critique issue de la loi du t de Student pour 764 ddl est 1.963074 soit 1.963 l'intervalle de confiance est donc sans doute [ 194.3357 ; 215.2590 ] soit, en arrondi, [ 194.336 ; 215.259 ]Si le test paramétrique dit que les longueurs ne sont pas significativement différentes alors que le test non paramétrique dit que les longueurs sont significativement différentes, que faut-il en conclure ? Tout d'abord qu'il faut être prudent[e]. Et ensuite essayer de voir ce qui explique les différences. La boite à moustaches précédente montre qu'il y a sans doute 2 longueurs extrêmes pour la DBDB et sans doute 4 longueurs extrêmes pour la LEAPDB.
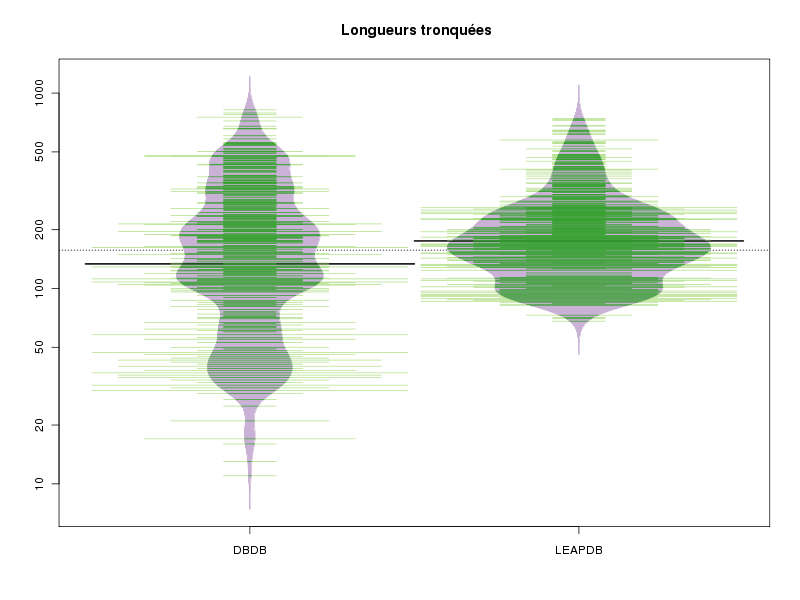
Même après avoir enlevé ces valeurs il n'y a pas accord entre les tests. Un beanplot fournit la réponse : il y a beaucoup plus de petites longueurs dans la DBDB.
# trions les longueurs lngLEAPDB <- sort( lngLEAPDB ) lngDBDB <- sort( lngDBDB ) # regardons les 10 dernières valeurs de lngLEAPDB cat("plus grandes valeurs dans lngLEAPDB\n"); print( tail(lngLEAPDB,n=10) ) # et de lngLEAPDB cat("plus grandes valeurs dans lngDBDB\n"); # cat est mieux que print... cat( tail(lngDBDB,n=10),"\n" ) # on enlève les 2 dernières valeurs de lngDBDB nbAoter <- 2 nbAgarder <- length(lngDBDB) - nbAoter lngDBDB_tr <- lngDBDB[ 1 : nbAgarder ] cat("nouvelles plus grandes valeurs dans lngDBDB\n"); # cat est mieux que print... cat( tail(lngDBDB_tr,n=10),"\n" ) # on enlève les valeurs supérieures à 1000 nbAoter <- length( which(lngLEAPDB>1000) ) nbAgarder <- length(lngLEAPDB) - nbAoter lngLEAPDB_tr <- lngLEAPDB[ 1 : nbAgarder ] cat("nouvelles plus grandes valeurs dans lngLEAPDB\n"); # cat est mieux que print... cat( tail(lngLEAPDB_tr,n=10),"\n" ) # et on recommence les comparaisons compMoyData("Longueurs tronquées","DBDB",lngDBDB_tr,"LEAPDB",lngLEAPDB_tr) print( wilcox.test(lngDBDB_tr,lngLEAPDB_tr) ) # essayons de comprendre graphiquement ce qui se passe : n1 <- length(lngDBDB_tr) n2 <- length(lngLEAPDB_tr) col_tr <- c( rep("blue",n1), rep("red",n2) ) base_tr <- c( rep("DBDB",n1), rep("LEAPDB",n2) ) lngs_tr <- c( lngDBDB_tr, lngLEAPDB_tr) idx <- order(lngs_tr) library(beanplot) png("complngtr1.png",width=800,height=600) boxplot(lngs_tr ~ base_tr, main=" Longueurs tronquées",col="yellow") dev.off() png("complngtr2.png",width=800,height=600) beanplot(lngs_tr ~ base_tr, main=" Longueurs tronquées", col = c("#CAB2D6", "#33A02C", "#B2DF8A"), border = "#CAB2D6") dev.off()plus grandes valeurs dans lngLEAPDB [1] 647 678 684 723 733 742 1236 1429 1509 1864 plus grandes valeurs dans lngDBDB 660 677 720 753 753 776 791 821 1045 1245 nouvelles plus grandes valeurs dans lngDBDB 656 659 660 677 720 753 753 776 791 821 nouvelles plus grandes valeurs dans lngLEAPDB 618 633 635 643 647 678 684 723 733 742 COMPARAISON DE MOYENNES (valeurs fournies) : Longueurs tronquées Variable NbVal Moyenne Variance Ecart-type Cdv DBDB 507 188.018 23865.828 154.486 82 % LEAPDB 761 197.940 12562.231 112.081 57 % Global 1268 193.972 17090.259 130.730 67 % Différence réduite : 1.2443 ; p-value 0.213726 Au seuil de 5 % soit 1.96, on ne peut pas rejeter l'hypothèse d'égalité des moyennes. En d'autres termes, il n'y a pas de différence significative entre les moyennes au seuil de 5 %. Voici ce qu'affiche la fonction t.test de R : Welch Two Sample t-test t = -1.2443, df = 853.255, p-value = 0.2137 alternative hypothesis: true difference in means is not equal to 0 95 percent confidence interval: -25.572201 5.728597 sample estimates: mean of x mean of y 188.0178 197.9396 A l'aide d'anova, on obtient Analysis of Variance Table Response: valeurs Df Sum Sq Mean Sq F value Pr(>F) groupes 1 29954 29954 1.7537 0.1856 Residuals 1266 21623404 17080 Wilcoxon rank sum test with continuity correction data: lngDBDB_tr and lngLEAPDB_tr W = 161272, p-value = 7.285e-07 alternative hypothesis: true location shift is not equal to 0
Les toutes petites valeurs rencontrées dans DBDB sont certainement incorrectes car il est peu probable qu'une dizaine d'acides aminés suffisent à faire une protéine...
> head(lngLEAPDB_tr,n=30) [1] 68 70 71 73 73 82 82 82 83 83 83 84 85 85 85 86 86 86 86 86 86 86 87 87 87 87 88 88 88 88 > head(lngDBDB_tr,n=30) [1] 11 13 16 17 17 17 17 21 21 21 25 27 29 29 30 30 30 30 30 30 31 31 31 32 32 32 32 32 32 33Celui de Husson, Lê et Pagès : analyse des données avec R.

Les fonctions présentées dans ce livre sont implémentées dans le package nommé FactoMiner.
Il existe aussi un package nommé ade4 très utile pour les analyses de données comme les ACP.
Ade4 est documenté à l'adresse http://pbil.univ-lyon1.fr/ade4/.
Il est possible que ces packages ne soient pas installés sur votre machine. Il convient donc de les installer par
install.packages("FactoMiner") install.packages("ade4")avant de les charger par
library(FactoMiner) library(ade4)Nos fonctions corCircle(), acpFacteur() et acp utilisent ces packages.