![]()
![]()
|
Partie Statistiques du cours de BioInformatiqueMaster BTV, UFR Sciences - Université d'AngersSolutions du TD numéro 2 (énoncés)
|
 Retour à la page principale de
(gH)
Retour à la page principale de
(gH)


Partie Statistiques du cours de BioInformatique
Master BTV, UFR Sciences - Université d'Angers
Solutions du TD numéro 2 (énoncés)
Pour comparer deux QT, on utilise un test d'hypothèse paramétrique ou non paramétrique suivant la normalité de la distribution des échantillons : pour des données normales non appariées, on utilise le test t de Student qui est équivalent à une ANOVA à un facteur ; pour des données normales appariées, on utilise le test t de Student adapté aux données appariées ; pour des données non normales non appariées, on utilise le test de Mann-Whitney ; pour des données non normales appariées, on utilise le test de Wilcox.
Voici le tableau résumé de ces divers cas pour comparer deux QT :
Données non appariées Données appariées Données normales test t de Student (ou anova) test t de Student adapté Données non normales test de Mann-Whitney test de Wilcox Pour comparer plus de deux QT, on utilise comme tests d'hypothèse paramétriques des anova, ancova, manova et autre mancova (donc pour des données normales) ; dans le cas non paramétrique, on utilise le test de Kruskal-Wallis pour des données non normales non appariées et le test de Friedman pour des données non normales appariées.
Voici le tableau résumé de ces divers cas pour comparer plus de deux QT :
Données non appariées Données appariées Données normales anova, ancova etc. anova, ancova etc. Données non normales test de Kruskal-Wallis test de Friedman En termes de graphiques, il est d'usage de tracer des boites à moustaches simultanées des QT, avec éventuellements des encoches (qui illustrent le test d'égalité des moyennes).
Il faut noter que, pour plus de deux QT, lorsque les tests d'hypothèses montrent des différences significatives entre les QT, on doit effectuer des tests post hoc pour déterminer quelles QT sont significativement différentes. De plus, ces comparaisons s'effectuent plutôt sur des groupes à l'intérieur d'une même QT que sur des QT différentes.
Pour étudier conjointement deux QL, on effectue un tri croisé puis un test d'indépendance du khi-deux sur ce tri croisé si les effectifs présents le permettent. En termes de graphiques, il est d'usage de tracer les histogrammes des effectifs des deux tris à plats et du tri croisé dans les deux sens afin de détecter d'éventuelles dépendances (changements de profils).
L'analyse conjointe de deux QT se fait impérativement après l'analyse séparée de chacune des deux QT. Ensuite, après l'étude de la normalité de chacune des distributions, on étudie, en cas de normalité, la corrélation linéaire à l'aide du coefficient de corrélation linéaire et, si le coefficient est significativement différent de zéro, on peut calculer les droites de régression. On doit aussi se poser la question de la causalité entre les QT ou la dépendance linéaire à une même troisième variable. S'il n'y a pas normalité, on s'intéressera à la corrélation des rangs (au sens de Spearman ou de Kendall).
Au niveau des graphiques, on tracera une QT en fonction de l'autre (diagramme de dispersion ou scatterplot) avec éventuellement la droite de régression linéaire.
Pour plus de deux QT, il faut dresser un tableau récapitulatif "bien trié" qui résume l'analyse séparée des QT puis calculer la matrice des coefficients de corrélation linéaire et s'intéresser aux plus forts coefficients. En terme de graphiques, un tracé systématique de tous les diagrammes deux à deux peut se révéler intéressant... On peut aussi réaliser une ACP (Analyse en Composantes Principales) mais sa lecture demande des compétences statistiques complémentaires.
Au lieu d'utiliser la fonction decritQT pour chaque règne, nous utiliserons la fonction decritQTparFacteurTexte de statgh.r comme suit :
source("statgh.r") lea2<-lit.dar("lea2.dar") attach(lea2) les4r <- as.character( sort(unique(reign2)) ) decritQTparFacteurTexte("LONGUEUR dans LEA2",length2,"aa","REGNE",reign2,les4r,TRUE) detach(lea2)dont voici les résultats
VARIABLE QT LONGUEUR dans LEA2 unité, : aa VARIABLE QL REGNE, labels : Bacteria Fungi Metazoa Viridiplantae N Moy Unite Ect Cdv Q1 Med Q3 EIQ Min Max Global 770 204.6325 aa 146.9473 72 128.5 168 234.8 106.2 68 1864 Bacteria 38 252.7105 aa 145.0865 57 157.8 195 290.5 132.8 102 647 Fungi 11 663.1818 aa 571.9613 86 245.5 417 960 714.5 138 1864 Metazoa 23 274.4348 aa 193.8123 71 102 182 376.5 274.5 70 742 Viridiplantae 698 192.4885 aa 111.9559 58 127.2 165.5 227 99.75 68 1429 Analysis of Variance Table Response: length2 Df Sum Sq Mean Sq F value Pr(>F) reign2 3 2615781 871927 47.669 < 2.2e-16 *** Residuals 766 14011214 18291 --- Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
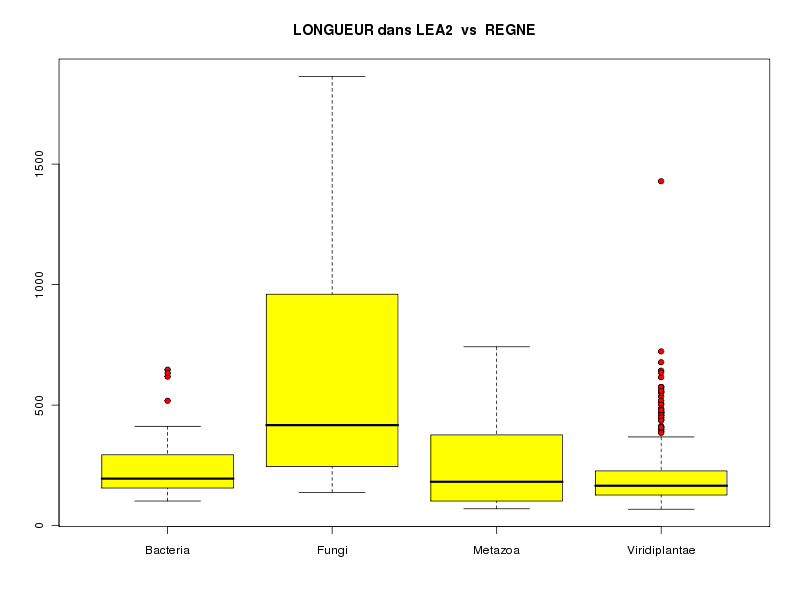
L'examen des calculs et graphiques mène à penser que Fungi est différent des autres règnes ; on se méfiera que sur le graphique, ce ne sont pas les moyennes mais les médianes qui sont affichées. A titre de comparaison, voici les boites à moustaches basées sur la médiane (à gauche) et basées sur la moyenne (à droite) fournies par le logiciel Statistica :
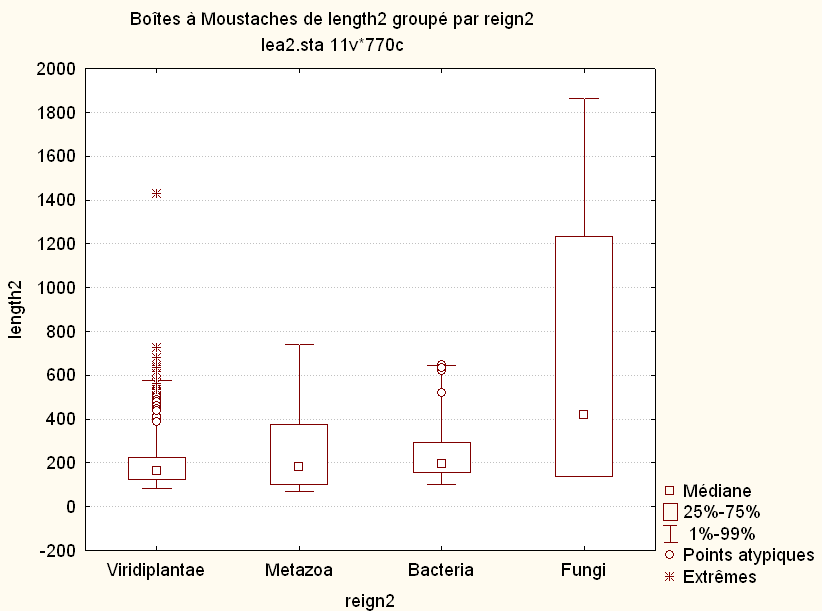
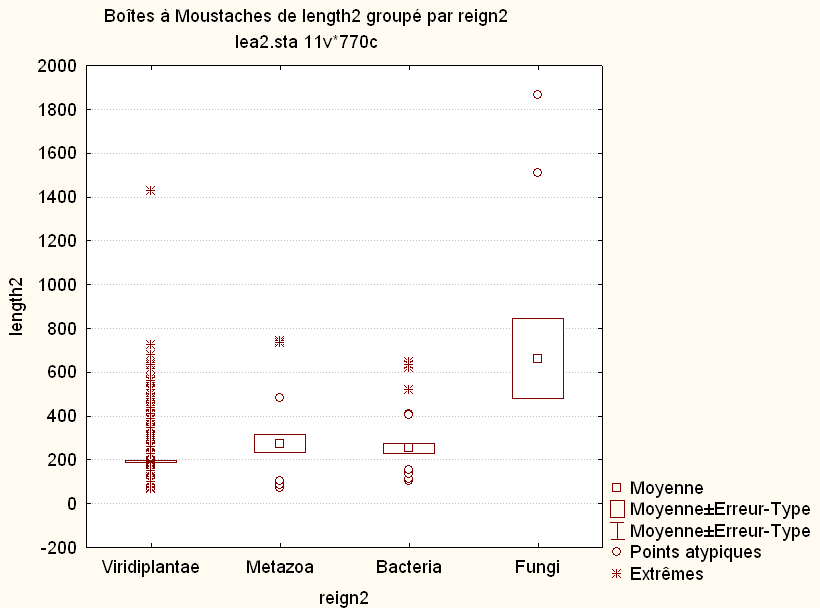
Au vu des histogrammes des variables bact et meta (non présentés ici), les données ne semblent pas normales. Comme de plus on a peu de données (moins de 50 pour chaque règne), on effectue un test (non paramétrique) de Mann-Whitney (nommé "test de Wilcox non apparié" par R) :
wilcox.test(bact,meta,paired=FALSE,exact=FALSE)Ce qu'affiche R permet de conclure qu'il n'y a pas de différence significative entre les longueurs de protéines au seuil de 5 % (car la "p-value" 0.7042 est supérieure à 0,05) :
Wilcoxon rank sum test with continuity correction data: bact and meta W = 463, p-value = 0.7042 alternative hypothesis: true location shift is not equal to 0
Le test d'hypothèse à utiliser est prop.test() mais nous lui préférons notre fonction compPourc() :
source("statgh.r") # les valeurs 557, 580 et 773 ont été calculées précédemment compPourc("CDD et PFAM non renseignés",557,773,580,773)dont voici les résultats
COMPARAISON DE POURCENTAGES CDD et PFAM non renseignés population A, 557 individus marqués sur 773 soit une proportion de 72.05692 % population B, 580 individus marqués sur 773 soit une proportion de 75.03234 % globalisation, 1137 individus marqués sur 1546 soit une proportion de 73.54463 % écart-réduit : 1.3261 ; "p-value" associée : 0.2046246 au seuil de 5 % soit 1.96, on peut accepter l'hypothèse d'égalité des pourcentages. En d'autres termes, il n'y a pas de différence significative entre les pourcentages au seuil de 5 %.L'exécution de l'instruction R suggérée par compPourc() en fin d'affichage est :
prop.test( c( 557 , 580 ) , c( 773 , 773 ) )et fournit comme résultats :
Exact binomial test data: c(557, 580) number of successes = 557, number of trials = 1137, p-value = 0.5141 alternative hypothesis: true probability of success is not equal to 0.5 95 percent confidence interval: 0.4604473 0.5193765 sample estimates: probability of success 0.4898857Ce qui permet de conclure, là encore, qu'il n'y a pas de différence significative entre les pourcentages au seuil de 5 %.
La question "Pourquoi y a-t-il autant de CDD (ou de PFAM) non renseignés ?" n'est pas une question de statistiques et aucune formule ni aucun logiciel ne peut y répondre. De plus, l'indication N/A indique seulement que notre lecture automatisée des fichiers Genpept du NCBI n'a pas permis de trouver de valeur pour CDD (ou pour PFAM). Ce qui peut s'interpréter de deux façons : soit la personne qui a déposé la séquence au NCBI n'a pas trouvé de CDD satisfaisant après les avoir tous passés en revue soit elle n'a même pas cherché.
Au lieu de l'instruction R nommée table qui sert aussi bien pour les tris à plat que les tris croisés, nous utiliserons nos fonctions decritQL et triCroise :
options(width=450) source("statgh.r") lea3<-lit.dar("lea3.dar") attach(lea3) labelCdd <- as.character(sort(unique(cdd3))) labelPfam <- as.character(sort(unique(pfam3))) decritQL("CDD renseigné",cdd3,labelCdd,TRUE) decritQL("PFAM renseigné",pfam3,labelPfam,TRUE) triCroise("PFAM renseigné",pfam3,labelPfam,"CDD renseigné",cdd3,labelCdd) detach(lea3)Nous obtenons alors :
> decritQL("CDD renseigné",cdd3,labelCdd,TRUE) QUESTION : CDD renseigné (ordre des modalités) 35153 66403 66647 66818 66886 67378 68499 68610 69026 69056 70305 84648 84797 85782 86004 88803 88917 91016 109320 Total Effectif 1 2 3 24 38 45 8 1 1 2 7 10 41 1 1 2 4 1 1 193 Frequence (en %) 1 1 2 12 20 23 4 1 1 1 4 5 21 1 1 1 2 1 1 103 QUESTION : CDD renseigné (par fréquence décroissante) 67378 84797 66886 66818 84648 68499 70305 88917 66647 66403 69056 88803 35153 68610 69026 85782 86004 91016 109320 Total Effectif 45 41 38 24 10 8 7 4 3 2 2 2 1 1 1 1 1 1 1 193 Frequence (en %) 23 21 20 12 5 4 4 2 2 1 1 1 1 1 1 1 1 1 1 103 > decritQL("PFAM renseigné",pfam3,labelPfam,TRUE) QUESTION : PFAM renseigné (ordre des modalités) 257 477 1442 1936 2496 2714 2987 3168 3242 3760 4927 5042 5478 5512 6830 Total Effectif 13 45 1 1 1 3 3 24 38 45 8 1 1 2 7 193 Frequence (en %) 7 23 1 1 1 2 2 12 20 23 4 1 1 1 4 103 QUESTION : PFAM renseigné (par fréquence décroissante) 477 3760 3242 3168 257 4927 6830 2714 2987 5512 1442 1936 2496 5042 5478 Total Effectif 45 45 38 24 13 8 7 3 3 2 1 1 1 1 1 193 Frequence (en %) 23 23 20 12 7 4 4 2 2 1 1 1 1 1 1 103 > triCroise("PFAM renseigné",pfam3,labelPfam,"CDD renseigné",cdd3,labelCdd) TRI CROISE DES QUESTIONS : PFAM renseigné (en ligne) CDD renseigné (en colonne) Effectifs 35153 66403 66647 66818 66886 67378 68499 68610 69026 69056 70305 84648 84797 85782 86004 88803 88917 91016 109320 257 0 0 0 0 0 0 0 0 0 0 0 10 0 0 0 2 0 0 1 477 0 0 0 0 0 0 0 0 0 0 0 0 41 0 0 0 4 0 0 1442 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 1936 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 2496 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 2714 1 2 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 2987 0 0 3 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 3168 0 0 0 24 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 3242 0 0 0 0 38 0 0 0 0 0 0 0 0 0 0 0 0 0 0 3760 0 0 0 0 0 45 0 0 0 0 0 0 0 0 0 0 0 0 0 4927 0 0 0 0 0 0 8 0 0 0 0 0 0 0 0 0 0 0 0 5042 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 5478 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 5512 0 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 6830 0 0 0 0 0 0 0 0 0 0 7 0 0 0 0 0 0 0 0 Valeurs en % du total 35153 66403 66647 66818 66886 67378 68499 68610 69026 69056 70305 84648 84797 85782 86004 88803 88917 91016 109320 TOTAL 257 0 0 0 0 0 0 0 0 0 0 0 5 0 0 0 1 0 0 1 7 477 0 0 0 0 0 0 0 0 0 0 0 0 21 0 0 0 2 0 0 23 1442 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 1 1936 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 1 2496 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 1 2714 1 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 2 2987 0 0 2 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 2 3168 0 0 0 12 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 12 3242 0 0 0 0 20 0 0 0 0 0 0 0 0 0 0 0 0 0 0 20 3760 0 0 0 0 0 23 0 0 0 0 0 0 0 0 0 0 0 0 0 23 4927 0 0 0 0 0 0 4 0 0 0 0 0 0 0 0 0 0 0 0 4 5042 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 1 5478 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 1 5512 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 1 6830 0 0 0 0 0 0 0 0 0 0 4 0 0 0 0 0 0 0 0 4 TOTAL 1 1 2 12 20 23 4 1 1 1 4 5 21 1 1 1 2 1 1 100
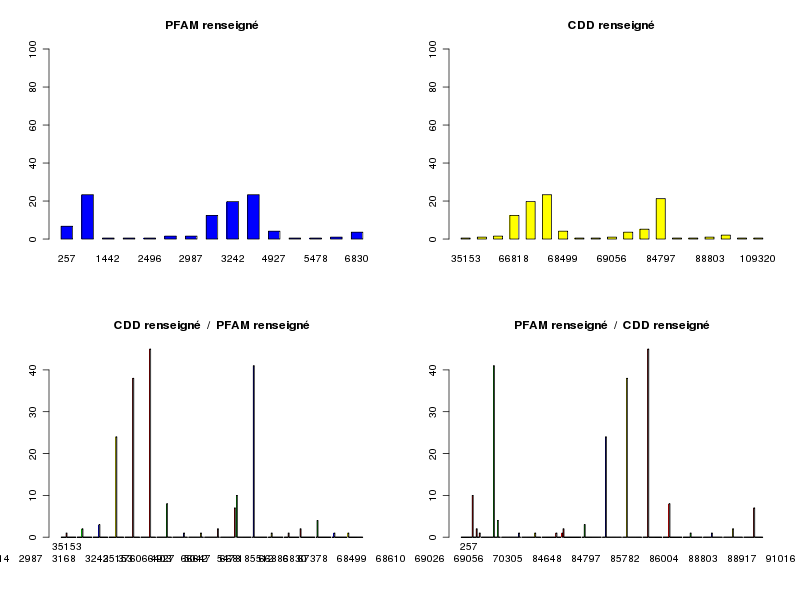
La lecture des tris à plats et du tri croisé montre que certains CDD et PFAM sont spécifiques à une protéine car de nombreux effectifs sont inférieurs à 5. Si on se limite aux effectifs supérieurs à 5, on obtient le tableau restreint suivant qui indique que CDD et PFAM "fonctionnent ensemble" :
66818 66886 67378 84648 84797 3168 24 0 0 0 0 3242 0 38 0 0 0 3760 0 0 45 0 0 257 0 0 0 10 0 477 0 0 0 0 41En toute rigueur, il n'est pas possible d'effectuer un test du khi-deux car de nombreux effectifs théoriques sont inférieurs à 5 :
CALCUL DU CHI-DEUX D'INDEPENDANCE ================================= TABLEAU DES DONNEES 257 477 1442 1936 2496 2714 2987 3168 3242 3760 4927 5042 5478 5512 6830 Total 35153 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 1 66403 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 2 66647 0 0 0 0 0 0 3 0 0 0 0 0 0 0 0 3 66818 0 0 0 0 0 0 0 24 0 0 0 0 0 0 0 24 66886 0 0 0 0 0 0 0 0 38 0 0 0 0 0 0 38 67378 0 0 0 0 0 0 0 0 0 45 0 0 0 0 0 45 68499 0 0 0 0 0 0 0 0 0 0 8 0 0 0 0 8 68610 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 1 69026 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 1 69056 0 0 0 0 0 0 0 0 0 0 0 0 0 2 0 2 70305 0 0 0 0 0 0 0 0 0 0 0 0 0 0 7 7 84648 10 0 0 0 0 0 0 0 0 0 0 0 0 0 0 10 84797 0 41 0 0 0 0 0 0 0 0 0 0 0 0 0 41 85782 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 1 86004 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 1 88803 2 0 0 0 0 0 0 0 0 0 0 0 0 0 0 2 88917 0 4 0 0 0 0 0 0 0 0 0 0 0 0 0 4 91016 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 1 109320 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 Total 13 45 1 1 1 3 3 24 38 45 8 1 1 2 7 193 VALEURS ATTENDUES et MARGES 257 477 1442 1936 2496 2714 2987 3168 3242 3760 4927 5042 5478 5512 6830 Total 35153 0.067 0.23 0.0052 0.0052 0.0052 0.016 0.016 0.12 0.20 0.23 0.041 0.0052 0.0052 0.010 0.036 1 66403 0.135 0.47 0.0104 0.0104 0.0104 0.031 0.031 0.25 0.39 0.47 0.083 0.0104 0.0104 0.021 0.073 2 66647 0.202 0.70 0.0155 0.0155 0.0155 0.047 0.047 0.37 0.59 0.70 0.124 0.0155 0.0155 0.031 0.109 3 66818 1.617 5.60 0.1244 0.1244 0.1244 0.373 0.373 2.98 4.73 5.60 0.995 0.1244 0.1244 0.249 0.870 24 66886 2.560 8.86 0.1969 0.1969 0.1969 0.591 0.591 4.73 7.48 8.86 1.575 0.1969 0.1969 0.394 1.378 38 67378 3.031 10.49 0.2332 0.2332 0.2332 0.699 0.699 5.60 8.86 10.49 1.865 0.2332 0.2332 0.466 1.632 45 68499 0.539 1.87 0.0415 0.0415 0.0415 0.124 0.124 0.99 1.58 1.87 0.332 0.0415 0.0415 0.083 0.290 8 68610 0.067 0.23 0.0052 0.0052 0.0052 0.016 0.016 0.12 0.20 0.23 0.041 0.0052 0.0052 0.010 0.036 1 69026 0.067 0.23 0.0052 0.0052 0.0052 0.016 0.016 0.12 0.20 0.23 0.041 0.0052 0.0052 0.010 0.036 1 69056 0.135 0.47 0.0104 0.0104 0.0104 0.031 0.031 0.25 0.39 0.47 0.083 0.0104 0.0104 0.021 0.073 2 70305 0.472 1.63 0.0363 0.0363 0.0363 0.109 0.109 0.87 1.38 1.63 0.290 0.0363 0.0363 0.073 0.254 7 84648 0.674 2.33 0.0518 0.0518 0.0518 0.155 0.155 1.24 1.97 2.33 0.415 0.0518 0.0518 0.104 0.363 10 84797 2.762 9.56 0.2124 0.2124 0.2124 0.637 0.637 5.10 8.07 9.56 1.699 0.2124 0.2124 0.425 1.487 41 85782 0.067 0.23 0.0052 0.0052 0.0052 0.016 0.016 0.12 0.20 0.23 0.041 0.0052 0.0052 0.010 0.036 1 86004 0.067 0.23 0.0052 0.0052 0.0052 0.016 0.016 0.12 0.20 0.23 0.041 0.0052 0.0052 0.010 0.036 1 88803 0.135 0.47 0.0104 0.0104 0.0104 0.031 0.031 0.25 0.39 0.47 0.083 0.0104 0.0104 0.021 0.073 2 88917 0.269 0.93 0.0207 0.0207 0.0207 0.062 0.062 0.50 0.79 0.93 0.166 0.0207 0.0207 0.041 0.145 4 91016 0.067 0.23 0.0052 0.0052 0.0052 0.016 0.016 0.12 0.20 0.23 0.041 0.0052 0.0052 0.010 0.036 1 109320 0.067 0.23 0.0052 0.0052 0.0052 0.016 0.016 0.12 0.20 0.23 0.041 0.0052 0.0052 0.010 0.036 1 Total 13.000 45.00 1.0000 1.0000 1.0000 3.000 3.000 24.00 38.00 45.00 8.000 1.0000 1.0000 2.000 7.000 193Toutefois notre fonction triCroise() permet de passer en revue les contributions au calcul de la distance du khi-deux : et montre bien que l'hypothèse d'indépendance ne peut pas être retenue en fournissant les plus fortes dépendances entre les modalités de CDD et PFAM pour des effectifs théoriques supérieurs à 5 :
Valeur du chi-deux 2702 Le chi-deux max (table) à 5 % est 290.0285 ; p-value 0 pour 252 degrés de liberte PLUS FORTES CONTRIBUTIONS AVEC SIGNE DE DIFFERENCE (EXTRAIT) Signe Valeur Pct CDD PFAM Ligne Colonne Obs Th + 124.482 4.61 % 66886 3242 5 9 38 7.5 + 113.492 4.20 % 67378 3760 6 10 45 10.5 + 103.404 3.83 % 84797 477 13 2 41 9.6 - 10.492 0.39 % 67378 477 6 2 0 10.5 - 9.560 0.35 % 84797 3760 13 10 0 9.6
Plutôt que d'utiliser deux fois decritQT(), une fois pour length et une fois pour mw, nous utiliserons notre fonction allQT qui fournit un affichage plus compact puis notre fonction anaLin :
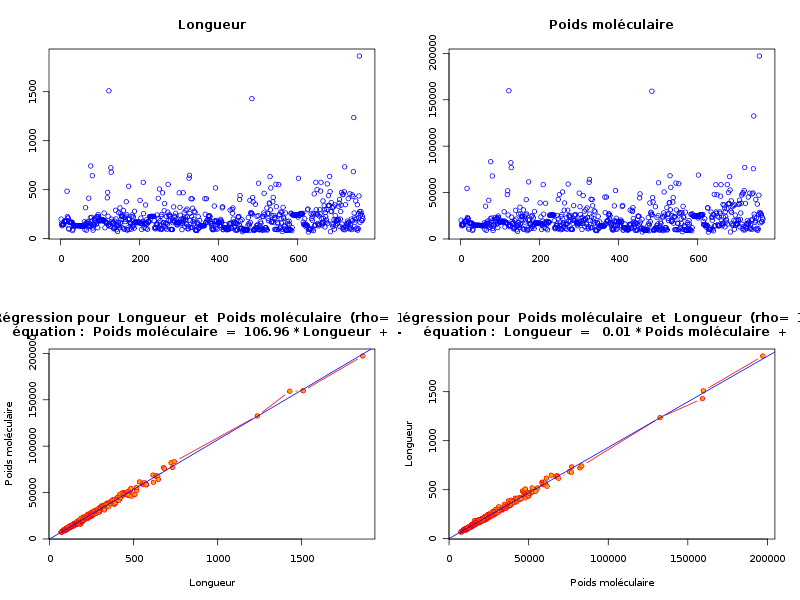
source("statgh.r") lea<-lit.dar("lea.dar") lengthmw <- lea[,c(1,7)] print(head(lengthmw)) allQT(lengthmw,"length mw","aa uma") attach(lea) anaLin("Longueur",length,"aa","Poids moléculaire",mw,"uma",TRUE) detach(lea)En voici les résultats
ANALYSE DES VARAIABLES QUANTITATIVES Par cdv décroissant Num Nom Taille Moyenne Unite Ecart-type Coef. de var. Minimum Maximum 2 mw 773 21838.965 uma 16041.950 73.46 % -9.000 197129.312 1 length 773 205.688 aa 148.596 72.24 % 68.000 1864.000 Par ordre d'entrée Num Nom Taille Moyenne Unite Ecart-type Coef. de var. Minimum Maximum 1 length 773 205.688 aa 148.596 72.24 % 68.000 1864.000 2 mw 773 21838.965 uma 16041.950 73.46 % -9.000 197129.312 Par moyenne décroissante Num Nom Taille Moyenne Unite Ecart-type Coef. de var. Minimum Maximum 2 mw 773 21838.965 uma 16041.950 73.46 % -9.000 197129.312 1 length 773 205.688 aa 148.596 72.24 % 68.000 1864.000 ANALYSE DE LA LIAISON LINEAIRE ENTRE Longueur ET Poids moléculaire coefficient de corrélation : 0.9922021 donc R2 = 0.984465 p-value associée : 0 équation : Poids moléculaire = 107.11 * Longueur - 193.30 équation : Longueur = 0.01 * Poids moléculaire + 4.97
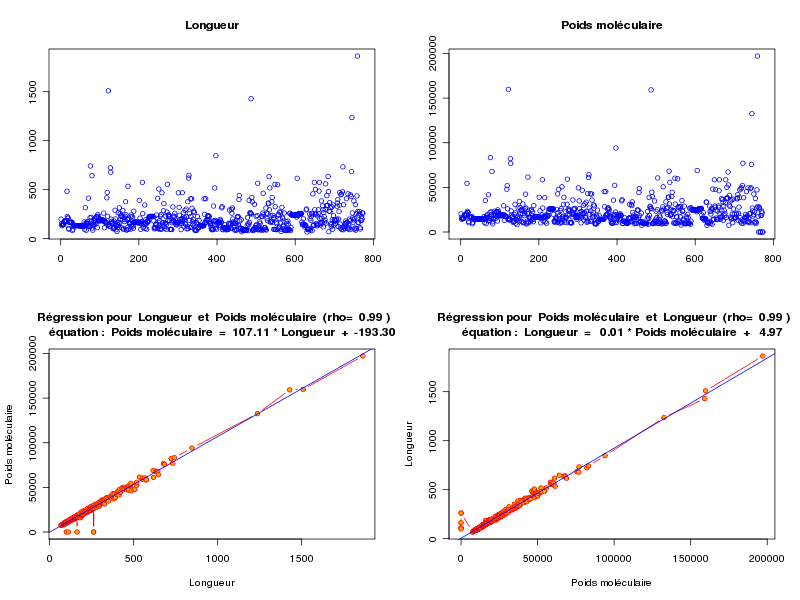
L'examen des calculs et graphiques montre qu'il y a une corrélation linéaire significative entre length et mw. Avec une longueur moyenne d'environ 206 aa et un écart-type d'environ 149 aa, soit un cdv de 72 %, les protéines semblent globalement assez homogènes au niveau de la longueur. La même remarque s'applique au poids moléculaire.
Remarque : sans nos fonctions, l'étude de la relation linéaire entre length et mw peut se faire sous R via
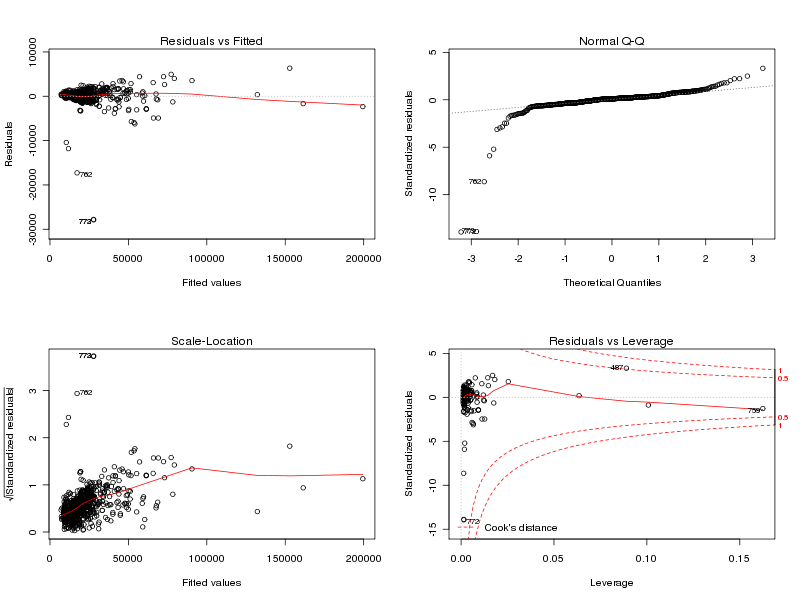
print(summary(lengthmw)) lg <- lea[,1] mw <- lea[,7] cat("Analyse de la corrélation : r = ",cor(lg,mw),"\n") print(cor.test(lg,mw)) an <- lm(mw~lg) print(an) print(anova(an)) par(mfrow=c(2, 2)) plot(an) par(mfrow=c(1,1))dont les résultats sont
length mw Min. : 68.0 Min. : -9 1st Qu.: 130.0 1st Qu.: 13961 Median : 168.0 Median : 17447 Mean : 205.7 Mean : 21839 3rd Qu.: 236.0 3rd Qu.: 25100 Max. :1864.0 Max. :197129 Analyse de la corrélation : r = 0.9922021 Pearson's product-moment correlation data: lg and mw t = 221.0406, df = 771, p-value < 2.2e-16 alternative hypothesis: true correlation is not equal to 0 95 percent confidence interval: 0.9910242 0.9932259 sample estimates: cor 0.9922021 Call: lm(formula = mw ~ lg) Coefficients: (Intercept) lg -193.3 107.1 Analysis of Variance Table Response: mw Df Sum Sq Mean Sq F value Pr(>F) lg 1 1.9584e+11 1.9584e+11 48859 < 2.2e-16 *** Residuals 771 3.0903e+09 4.0082e+06 --- Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' '
Et un tracé élémentaire de mw en fonction de lg aurait été obtenu par :
plot(lg,mw) plot(lg,mw,main=" tracé de mw en fonction de lg") abline(lm(mw~lg),col="red")
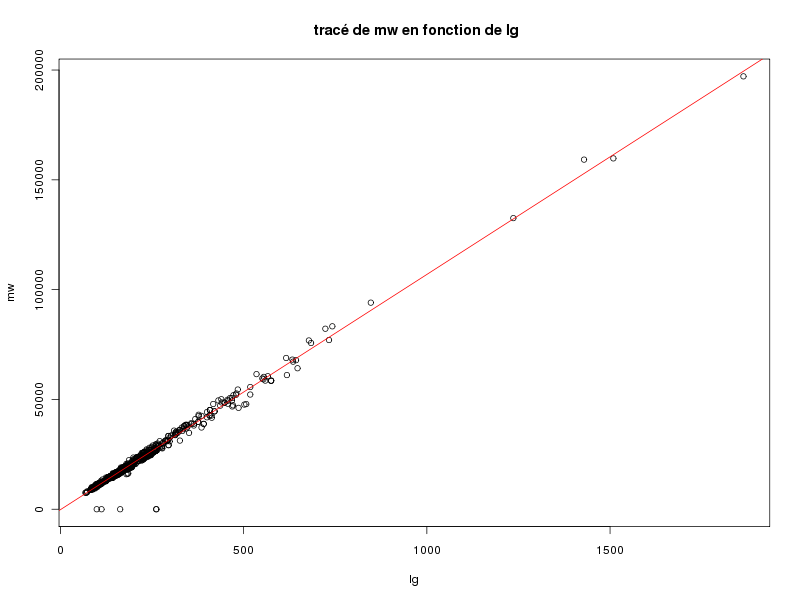
On pourra s'étonner du minimum -9 pour mw (qui est un "code" pour indiquer que la donnée est manquante) et remarquer que la masse moléculaire moyenne s'obtient globalement en prenant 107 fois la longueur et en retirant 193 (chaque aa ajoutant 107 à cette masse). La causalité ici est particulièrement évidente : plus la longueur augmente, plus la masse moléculaire augmentera, la réciproque n'étant pas vraie : peu de "gros aa" (Arginine et Tryptophane) augmentent sensiblement la masse moléculaire mais pas la longueur. Par contre, la linéarité n'était pas "évidente" et surtout pas avec un tel coefficient de corrélation linéaire.
Puisque le fichier des données contient des données manquantes (code -9), il faut les supprimer. Le fichier lea4.dar contient le fichier correct des données. Il faut recommencer l'étude de régression. Voici donc les "vrais" résultats qui, heureusement, changent peu des précédents :
source("statgh.r") lea<-lit.dar("lea4.dar") lengthmw <- lea[,c(1,7)] print(head(lengthmw)) allQT(lengthmw,"length mw","aa uma") attach(lea) anaLin("Longueur",length4,"aa","Poids moléculaire",mw4,"uma",TRUE) detach(lea)ANALYSE DES VARAIABLES QUANTITATIVES Par cdv décroissant Num Nom Taille Moyenne Unite Ecart-type Coef. de var. Minimum Maximum 2 mw 765 21858.953 uma 15795.787 72.26 % 7521.360 197129.312 1 length 765 204.797 aa 147.303 71.93 % 68.000 1864.000 Par ordre d'entrée Num Nom Taille Moyenne Unite Ecart-type Coef. de var. Minimum Maximum 1 length 765 204.797 aa 147.303 71.93 % 68.000 1864.000 2 mw 765 21858.953 uma 15795.787 72.26 % 7521.360 197129.312 Par moyenne décroissante Num Nom Taille Moyenne Unite Ecart-type Coef. de var. Minimum Maximum 2 mw 765 21858.953 uma 15795.787 72.26 % 7521.360 197129.312 1 length 765 204.797 aa 147.303 71.93 % 68.000 1864.000 Matrice des corrélations length mw length 1.000 mw 0.997 1.000 Meilleure corrélation 0.9974722 pour mw et length Formules length = 0.009 * mw + 1.468 et mw = 106.962 * length -46.679 ANALYSE DE LA LIAISON LINEAIRE ENTRE Longueur ET Poids moléculaire coefficient de corrélation : 0.9974722 donc R2 = 0.9949508 p-value associée : 0 équation : Poids moléculaire = 106.96 * Longueur - 46.68 équation : Longueur = 0.01 * Poids moléculaire + 1.47