![]()
![]()
|
Partie Statistiques du cours de BioInformatiqueMaster BTV, UFR Sciences - Université d'AngersEnoncés du TD numéro 4 (solutions)
|
 Retour à la page principale de
(gH)
Retour à la page principale de
(gH)


Partie Statistiques du cours de BioInformatique
Master BTV, UFR Sciences - Université d'Angers
Enoncés du TD numéro 4 (solutions)
Donner la moyenne et l'écart des variables V1, V2, V3, V4 et V5 si on exécute les instructions R suivantes :
source("statgh.r") lea4 <- lit.dar("lea4.dar") lng <- lea4$length4 m <- mean(lng) s <- sd(lng) V1 <- lng V2 <- lng - m V3 <- lng/s V4 <- lng/s - m V5 <- (lng-m)/sOn dispose d'une variable QT nommée X de moyenne m et d'écart s ; comment faut-il choisir a et b pour que la variable Y définie par Y = a.X + b ait une moyenne de 0 et un écart-type de 1 ? Ecrire des fonctions R pour ces calculs de "normalisation".
Après avoir parcouru rapidement l'article bénomyl, pouvez-vous dire ce que contient le gros fichier (15 589 lignes) nommé 20min_Beno_Cy5_DMSO_Cy3.gpr dont un extrait est 20min_Beno_Cy5_DMSO_Cy3.mini ? Au passage, merci à Gaëlle Lelandais pour ces données.
A quoi correspond le suffixe gpr de ce fichier ? Comment lire ce fichier en R ? Donner la signification de Gb, Gf, Rb et Rf.
Après avoir lu la page d'aide sur les données swirl, expliquez ce que contient l'objet swirl disponible en R par data(swirl) lorsque le "package" marray (qui fait partie de la suite logicielle Bionconductor) est installé.
Quelles sont les étapes de la préparation statistique des données pour une puce à ADN ?
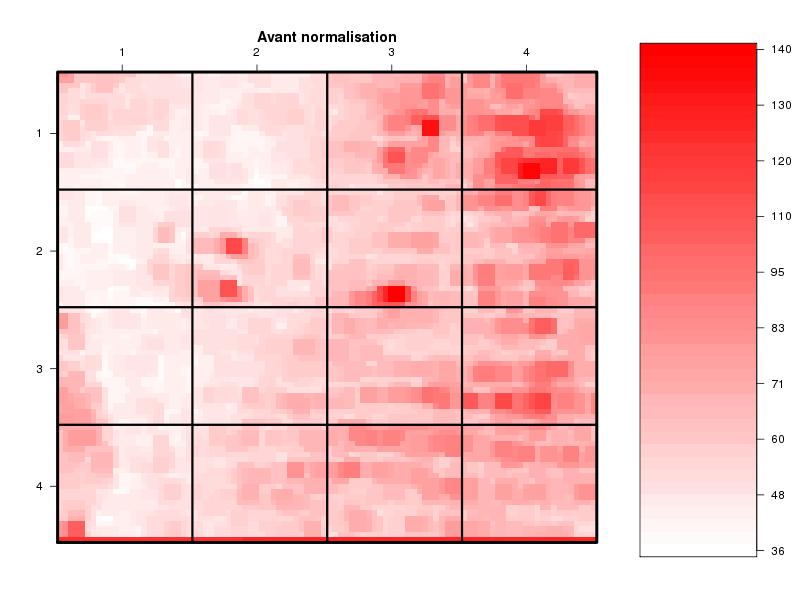
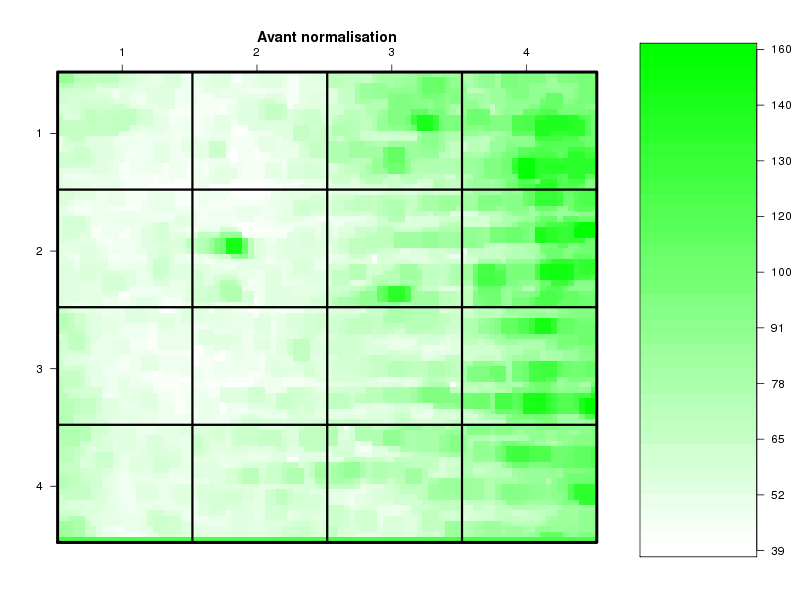
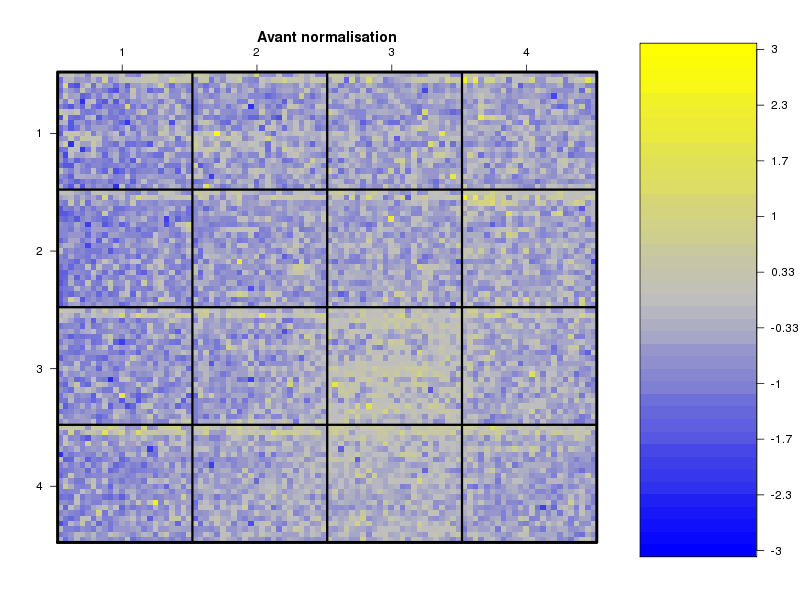
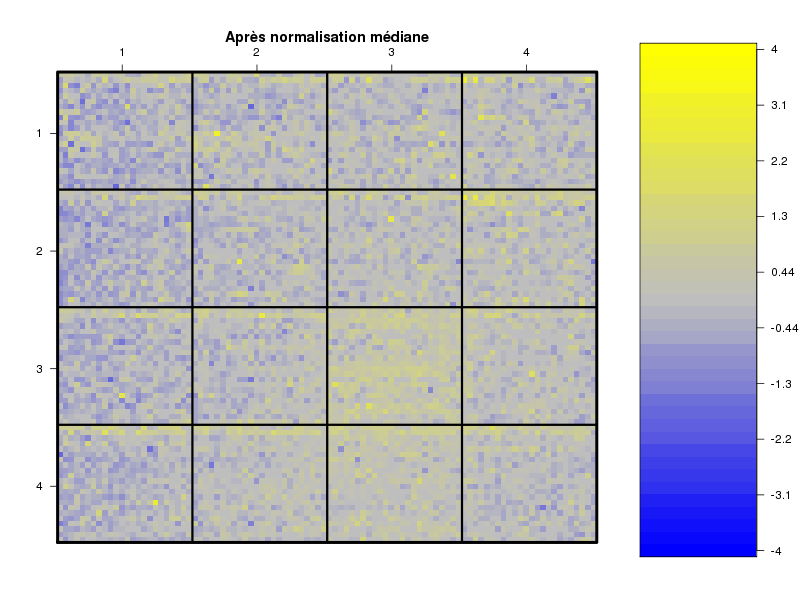
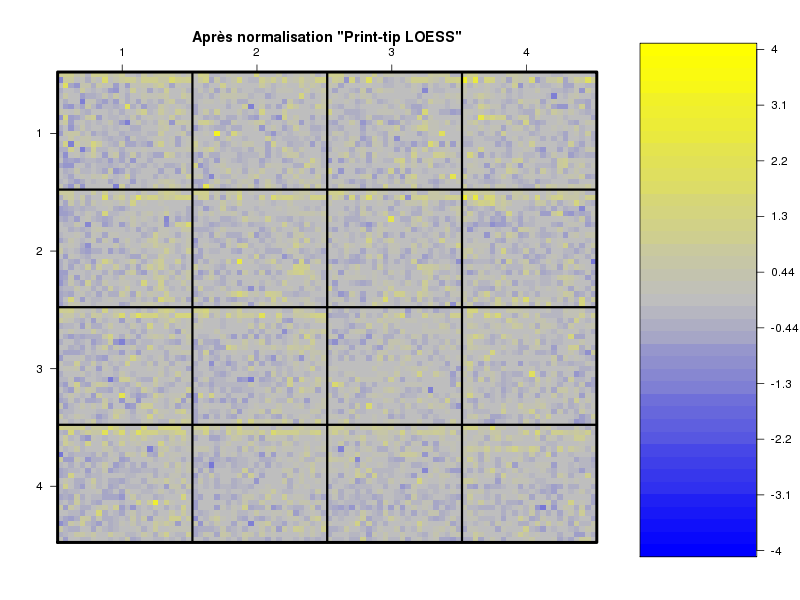
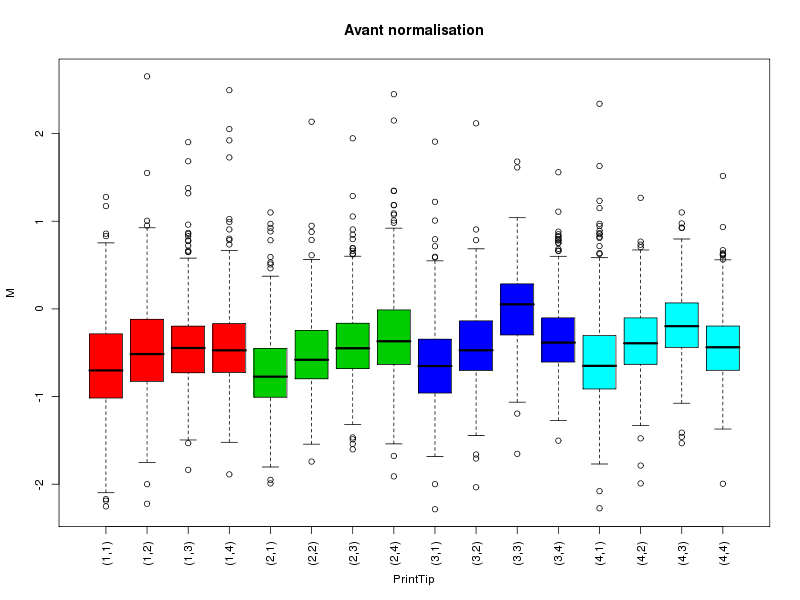
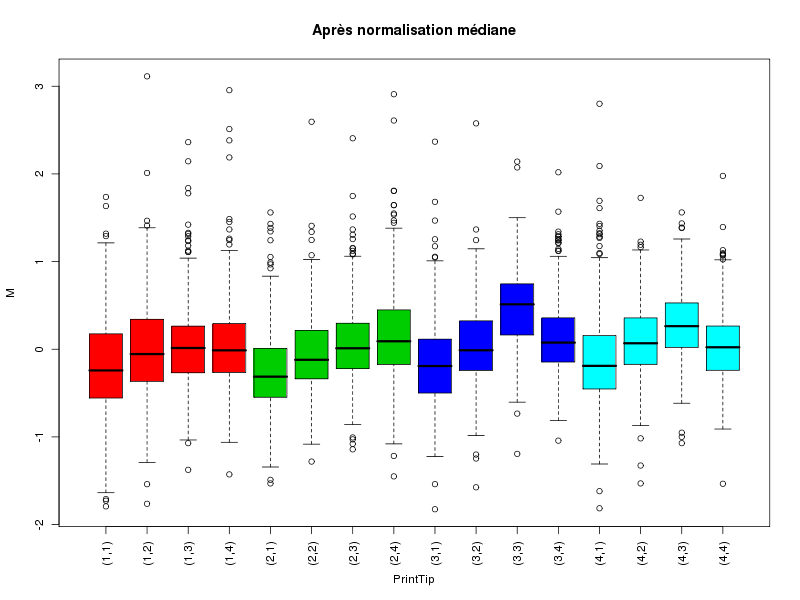
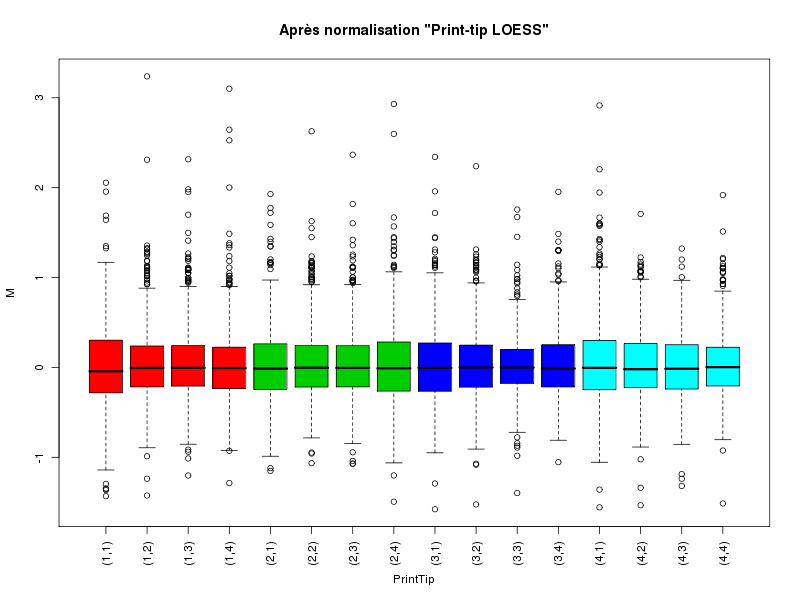
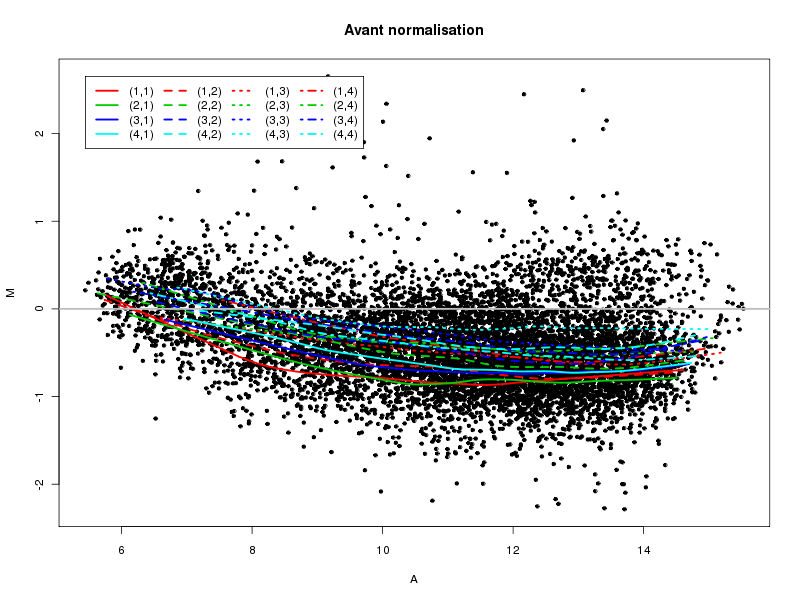
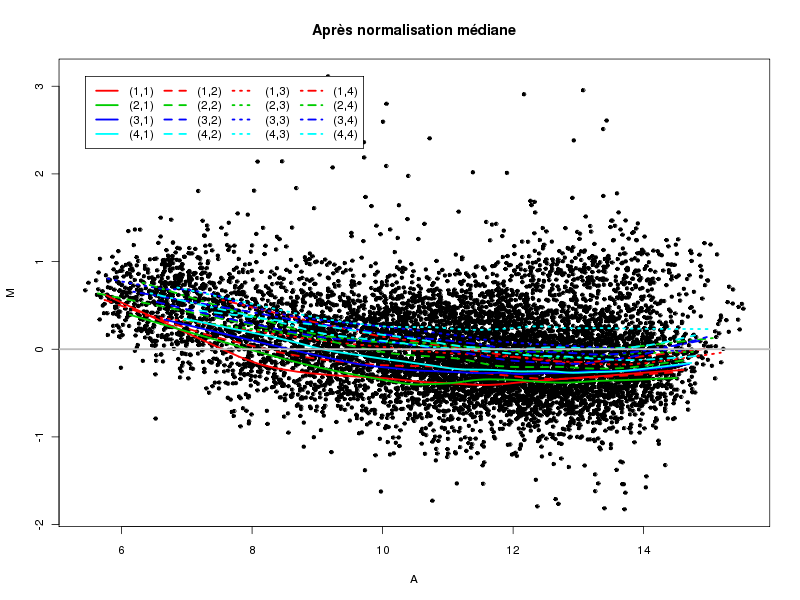
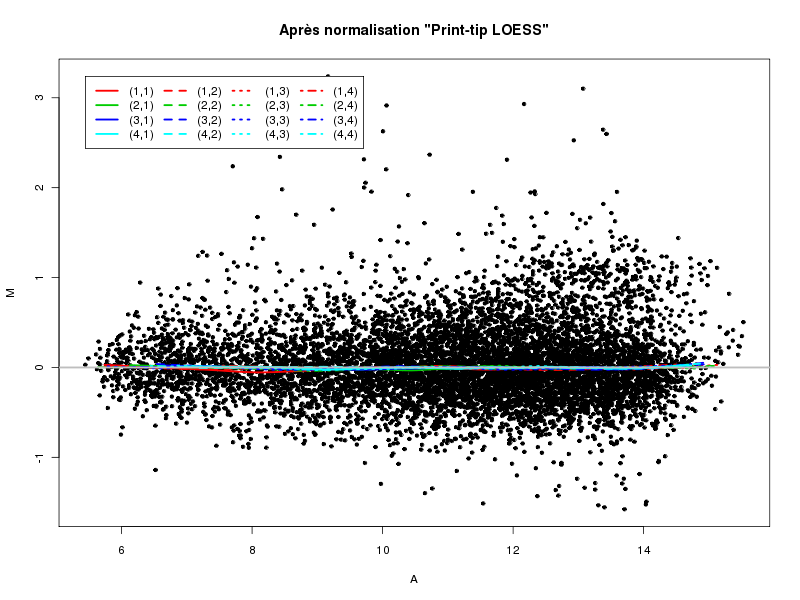
Commenter les instructions R suivantes (restructurées) fournies par jvanheld dans le cours microarrays à la rubrique "Normalization". On pourra s'inspirer des graphiques fournis.
## load the marray library library(marray) ## load the demo data set data(swirl) ## select a single chip for saving time (LOESS costs a lot of time) swirl.3 <- swirl[,3] ## normalize the swirl data set swirl.norm.med <- maNorm(swirl.3,norm="median") ## Print-tip LOESS normalization: ## BEWARE: THIS CAN BE TIME CONSUMING. swirl.norm.printTipLoess <- maNorm(swirl.3,norm="printTipLoess") ################################################################### swimg <- function(grf,obj,tit,opt) { png(grf,width=800,height=600) if (opt==0) { maImage(obj,main=tit) } if (opt==1) { maImage(obj,main=tit,x="maRb") } if (opt==2) { maImage(obj,main=tit,x="maGb") } if (opt==3) { maBoxplot(obj,main=tit) } if (opt==4) { maPlot(obj,main=tit) } dev.off() } ; # fin fonction swimg ################################################################### tit1 = "Avant normalisation" ; tit2 = "Après normalisation médiane" ; tit3 = "Après normalisation \"Print-tip LOESS\"" ; ## Images of the 3rd chip of the raw, ## median-normalized and loess-normalized data swimg("vh1a.png",swirl.3,tit1,0) swimg("vh1b.png",swirl.norm.med,tit2,0) swimg("vh1c.png",swirl.norm.printTipLoess,tit3,0) ## Red and green background of the 3rd chip of the raw data swimg("vh2ar.png",swirl.3,tit1,1) swimg("vh2ag.png",swirl.3,tit1,2) ## boxplots swimg("vh3a.png",swirl.3 ,tit1,3) swimg("vh3b.png",swirl.norm.med ,tit2,3) swimg("vh3c.png",swirl.norm.printTipLoess,tit3,3) # MA plots swimg("vh4a.png",swirl.3 ,tit1,4) swimg("vh4b.png",swirl.norm.med ,tit2,4) swimg("vh4c.png",swirl.norm.printTipLoess ,tit3,4)
Excel est-il un logiciel statistique ? Quels logiciels statistiques sont disponibles sur le marché ? Pour des analyse générales ? Pour les puces à ADN ? Sont-ils gratuits, payants ?
Expliquer pourquoi le logiciel R est sans doute le bon choix de logiciel statistique pour vous, étudiant(e) de bioinformatique.
Peut-on utiliser des interfaces (GUI) et des menus pour R autres que Rcmdr ?
Que sont les taskviews du CRAN ? Qu'est-ce que BioConductoR ? et seqinr ?
Quel livre traite de R et de phylogénie ? Installez le package ape et exécutez le code R suivant. Qu'en déduisez-vous ?
library(ape) # si install.packages("ape",dependencies=TRUE) a été exécuté example(plot.phylo) example(plot.mst) example(plot.multiPhylo) example(plot.popsize) example(plot.prop.part) example(plot.skyline)Comment conclure ces 4 séances ?